School on Scientific Data Analysis, 25-28 November 2019 Scuola Normale





























































78 Slides6.17 MB
School on Scientific Data Analysis, 25-28 November 2019 Scuola Normale Superiore Data Analysis in Next Generation Sequencing Paolo Aretini Senior Researcher Fondazione Pisana per la Scienza
Outline Introduction Bioinformatics in NGS data analysis 1. 2. 3. 4. Basics: terminology, data formats, etc. Data Analysis Pipeline Sequence Quality Control and preprocessing Sequence mapping DNA-Seq data analysis 1. Post-alignment processing 2. Variant Calling 3. Variant Annotation, Filtration, Prioritization 4. NGS and rare diseases Summary
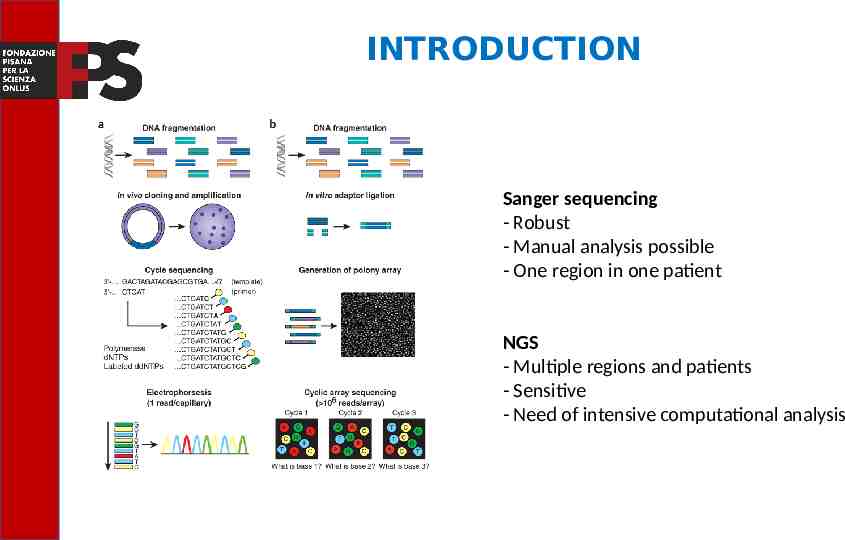
INTRODUCTION Next generation sequencing (NGS) is the set of nucleic acid sequencing technologies that have in common the ability to sequencing, in parallel, millions of DNA fragments. These technologies have marked a revolutionary turning point in the possibility of characterizing large genomes compared to the first generation DNA sequencing method (Sanger sequencing), because of the potential to produce, in a single analysis session, a quantity of genetic information millions of times larger.
INTRODUCTION The Sanger method was used as part of the "Human Genome Project" for the complete sequencing of the first human genome; this objective was achieved in 2003, after 13 years of work and at an estimated cost of 2.7 billion dollars. Today sequencing the genome costs 14 thousand times less, now it can be done with about 1000 dollars in a few days. This latest result highlights the rapid evolution in the field of next generation sequencing technologies.
INTRODUCTION Sanger sequencing - Robust - Manual analysis possible - One region in one patient NGS - Multiple regions and patients - Sensitive - Need of intensive computational analysis
INTRODUCTION On the market there are two producers of sequencing machine, Illumina and Thermofisher. llumina produces sequencers able of generating a greater amount of data (6 billion reads)
INTRODUCTION Basic NGS Workflow While the sequencing run is the same for each type of investigation, the sample preparation and data analysis are application specific.
INTRODUCTION NGS technologies are used for many applications: genetic variant discovery by Whole Genome Sequencing (WGS) or Whole Exome Sequencing (WES, genome encoding regions); transcriptome profiling of cells, tissues or organisms; many more applications (alternative splicing, identification of epigenetic markers; ChIP-Seq).
INTRODUCTION Bioinformatics Challenges in NGS Data Analysis The application of NGS techniques required additional Information Technology resources. “Big Data” It’s not possible to do ‘business as usual’ with familiar tools Manage, analyze, store and transfer huge files needed Need for powerful computers and expertise Informatics groups must manage compute clusters Algorithms and software are required and often time they are open source Unix/Linux based. Collaboration of IT experts, bioinformaticians and biologists
Outline Introduction Bioinformatics in NGS data analysis 1. 2. 3. 4. Basics: terminology, data formats, general workflow etc Data Analysis Pipeline Sequence QC and preprocessing Sequence mapping DNA-Seq data analysis 1.Post-alignment processing 2. Variant Calling 3. Variant Annotation, Filtration, Prioritization 4. NGS and rare diseases Summary
Terminology What is bioinformatics? Broad term: From AI to biostatistics Here: Computational analysis of NGS data Giving clinical significance to hundreds of genetic alterations
Terminology Genetic variant: An alteration in the most common DNA nucleotide sequence. The term variant can be used to describe an alteration that may be benign, pathogenic, or of unknown significance. Single-nucleotide variant: a single-nucleotide variant (SNV) is a variation in a single nucleotide without any limitations of frequency and may arise in germline or somatic cells. Indels: insertion–deletion mutations (indels) refer to insertion and/or deletion of nucleotides into genomic DNA. Indels are important in clinical next-generation sequencing (NGS), as they are implicated as the driving mechanism underlying many constitutional and oncologic diseases. Whole genome sequencing: (also known as WGS) is the process of determining the complete DNA sequence of an organism's genome at a single time. Exome sequencing: also known as whole exome sequencing (WES), is a genomic technique for sequencing all of the protein-coding region of genes in a genome (known as the exome)
Terminology Paired-End Sequencing: Both end of the DNA fragment is sequenced, allowing highly precise alignment. Quality Score: Each called base comes with a quality score which measures the probability of base call error. Mapping: Align reads to reference genome to identify their origin. Duplicate reads: Reads that are identical. Can be identified after mapping.
List of file formats in NGS data analysis FASTA – The FASTA file format, for sequence data. Sometimes also given as FNA or FAA (Fasta Nucleic Acid or Fasta Amino Acid). FASTQ – The FASTQ file format, for sequence data with quality. Raw data from sequencer. SAM – Sequence Alignment/Map format, in which the results of the 1000 Genomes Project will be released. BAM – Binary compressed SAM format. VCF – Variant Call Format, a standard created by the 1000 Genomes Project that lists the genetic variants generated by an NGS run.
Outline Introduction Bioinformatics in NGS data analysis 1. 2. 3. 4. Basics: terminology, data formats, general workflow etc Data Analysis Pipeline Sequence QC and preprocessing Sequence mapping DNA-Seq data analysis 1. Post-alignment processing 2. Variant Calling 3. Variant Annotation, Filtration, Prioritization 4. NGS and rare diseases Summary
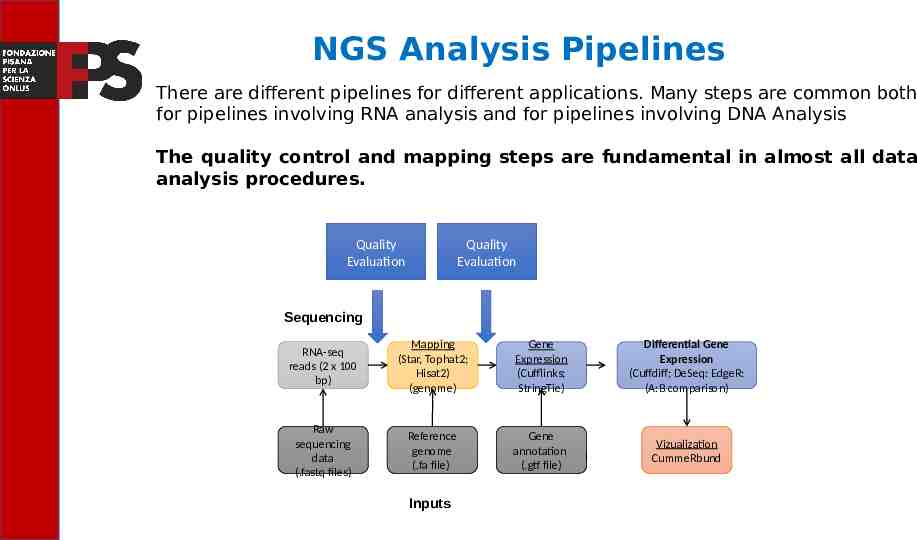
NGS Analysis Pipelines There are different pipelines for different applications. Many steps are common both for pipelines involving RNA analysis and for pipelines involving DNA Analysis The quality control and mapping steps are fundamental in almost all data analysis procedures. Quality Evaluation Quality Evaluation Sequencing Read alignment Transcript compilation RNA-seq reads (2 x 100 bp) Mapping (Star, Tophat2; Hisat2) (genome) Gene Expression (Cufflinks; StringTie) Differential Gene Expression (Cuffdiff; DeSeq: EdgeR: (A:B comparison) Raw sequencing data (.fastq files) Reference genome (.fa file) Gene annotation (.gtf file) Vizualization CummeRbund Inputs Differential expression
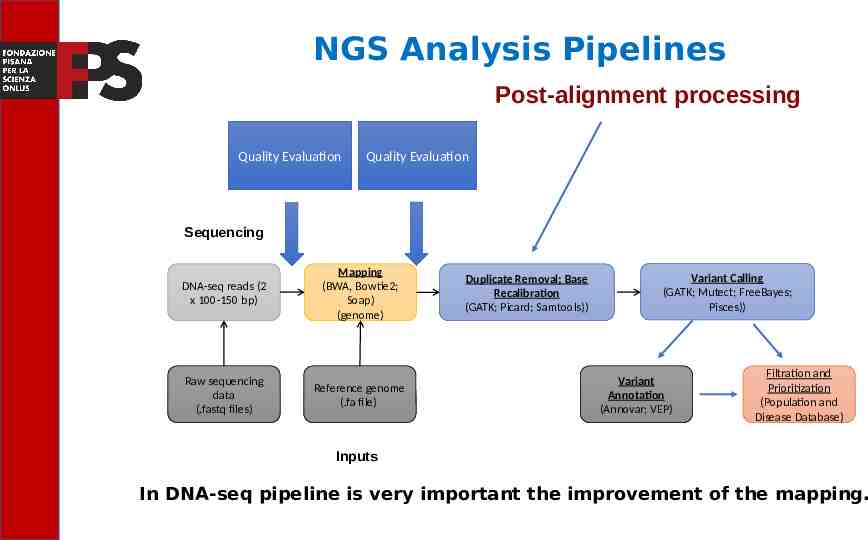
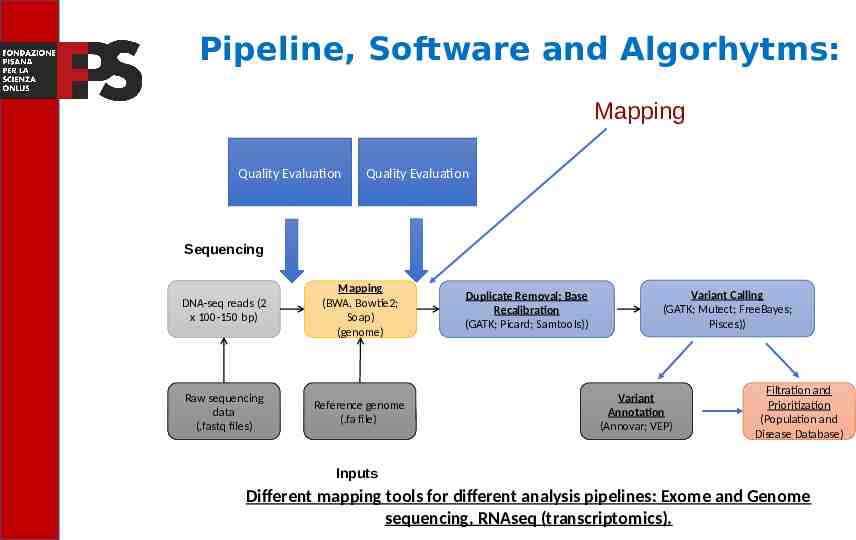
NGS Analysis Pipelines Post-alignment processing Quality Evaluation Quality Evaluation Sequencing Read alignment DNA-seq reads (2 x 100-150 bp) Mapping (BWA, Bowtie2; Soap) (genome) Raw sequencing data (.fastq files) Reference genome (.fa file) Transcript compilation Duplicate Removal; Base Recalibration (GATK; Picard; Samtools)) Differential expression Variant Calling (GATK; Mutect; FreeBayes; Pisces)) Variant Annotation (Annovar; VEP) Filtration and Prioritization (Population and Disease Database) Inputs In DNA-seq pipeline is very important the improvement of the mapping.
Outline Introduction Bioinformatics in NGS data analysis 1. 2. 3. 4. Basics: terminology, data formats, general workflow etc Data Analysis Pipeline Sequence QC and preprocessing Sequence mapping DNA-Seq data analysis 1. Post-alignment processing 2. Variant Calling 3. Variant Annotation, Filtration, Prioritization 4. NGS and rare diseases Summary
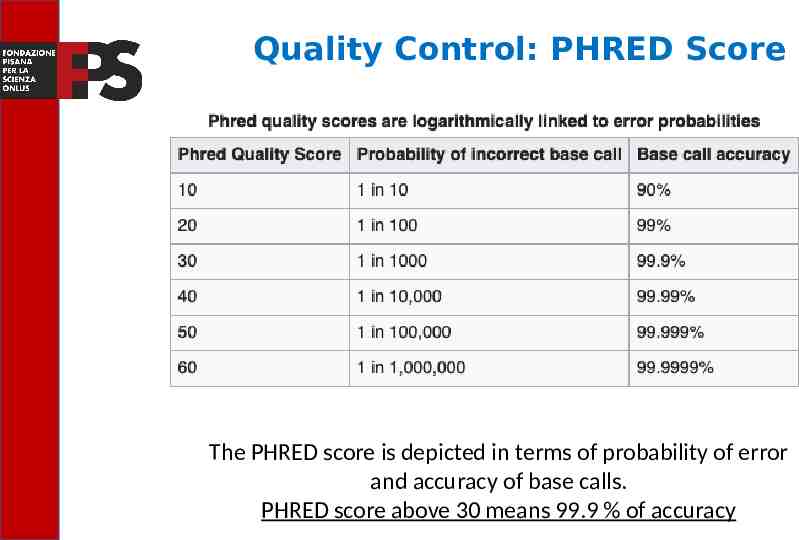
FASTQ Files The raw data from a sequencing machine are most widely provided as FASTQ files, which include sequence information, similar to FASTA files, but additionally contain further information, including sequence quality information. A FASTQ file consists of blocks, corresponding to reads, and each block consists of four elements in four lines. The last line encodes the quality score for the sequence in line 2 in the form of ASCII characters. The byte representing quality runs (lowest quality; '!' in ASCII; highest quality; ' ' in ASCII). The PHRED score is the most used scoring system and represents the probability, on a logarithmic scale, that a base is misread.
Quality Control: PHRED Score The PHRED score is depicted in terms of probability of error and accuracy of base calls. PHRED score above 30 means 99.9 % of accuracy
Quality Conytrol: Why? On the basis of the information contained in the fastq files it is possible to carry out a quality control and possibly improve the raw data to avoid errors in the downstream analysis. By performing QC at the beginning of the analysis, chances encountering any contamination, bias, error, and missing data are minimized.
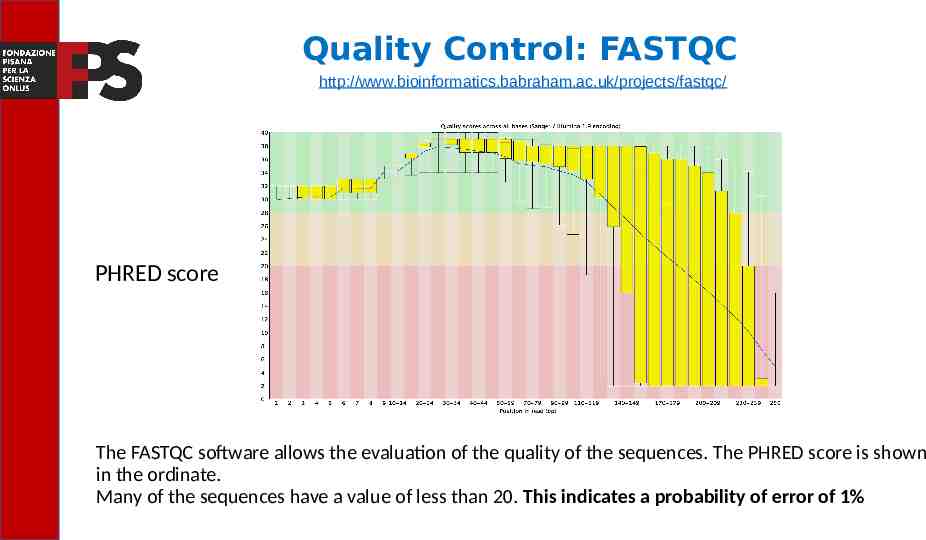
Quality Control: FASTQC http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ PHRED score The FASTQC software allows the evaluation of the quality of the sequences. The PHRED score is shown in the ordinate. Many of the sequences have a value of less than 20. This indicates a probability of error of 1%
FASTQ Files Tools for FASTQ manipulation an QC improving
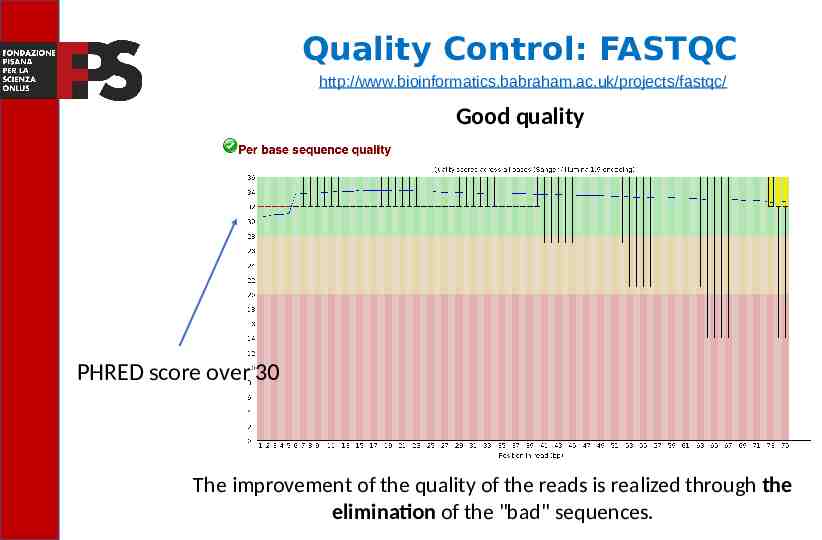
Quality Control: FASTQC http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ Good quality PHRED score over 30 The improvement of the quality of the reads is realized through the elimination of the "bad" sequences.
Outline Introduction Bioinformatics in NGS data analysis 1. 2. 3. 4. Basics: terminology, data formats, general workflow etc Data Analysis Pipeline Sequence QC and preprocessing Sequence mapping DNA-Seq data analysis 1. Improving the quality and robustness of mapping 2. Variant Calling 3. Variant Annotation, Filtration, Prioritization 4. NGS and rare diseases Summary
Pipeline, Software and Algorhytms: Mapping Quality Evaluation Quality Evaluation Sequencing Read alignment DNA-seq reads (2 x 100-150 bp) Mapping (BWA, Bowtie2; Soap) (genome) Raw sequencing data (.fastq files) Reference genome (.fa file) Transcript compilation Duplicate Removal; Base Recalibration (GATK; Picard; Samtools)) Differential expression Variant Calling (GATK; Mutect; FreeBayes; Pisces)) Variant Annotation (Annovar; VEP) Filtration and Prioritization (Population and Disease Database) Inputs Different mapping tools for different analysis pipelines: Exome and Genome sequencing, RNAseq (transcriptomics).
Mapping Mapping has fastq files as input and produces SAM files. Factors influencing mapping: Read length Sequencing libraries: single-end and paired-end sequencing Some pitfalls: sequencing errors, low quality reads, duplicated reads.
Mapping SAM Format The Sequence Alignment/Map (SAM) format is a generic alignment format for storing read alignments against reference sequences. It was firstly introduced by the 1000 Genomes Project Consortium to release the alignments performed. The SAM format consists of one header section and one alignment section. The header section contains information about the quality of the mapping, information about the instruments used and about the tools used.
Mapping BAM Format To improve the performance, 1000 genomes project consortium designed a companion format Binary Alignment/Map (BAM), which is the binary representation of SAM and keeps exactly the same information as SAM. BAM is compressed by the BGZF library, a generic library specifically developed to achieve fast random access in a zlib-compatible compressed file. BAM files can be sorted by chromosomal coordinates. This procedure allows for indexing the BAM. Index sorted alignment enables to efficiently retrieve all reads aligning to a locus.
Mapping SAMtools software package Samtools is a software that is used to manipulate SAM/BAM files and is one of the most used tools in the analysis of NGS data. It is able to convert from other alignment formats, sort and merge alignments, remove PCR duplicates, call SNPs and short indel variants, and show alignments in a text-based viewer.
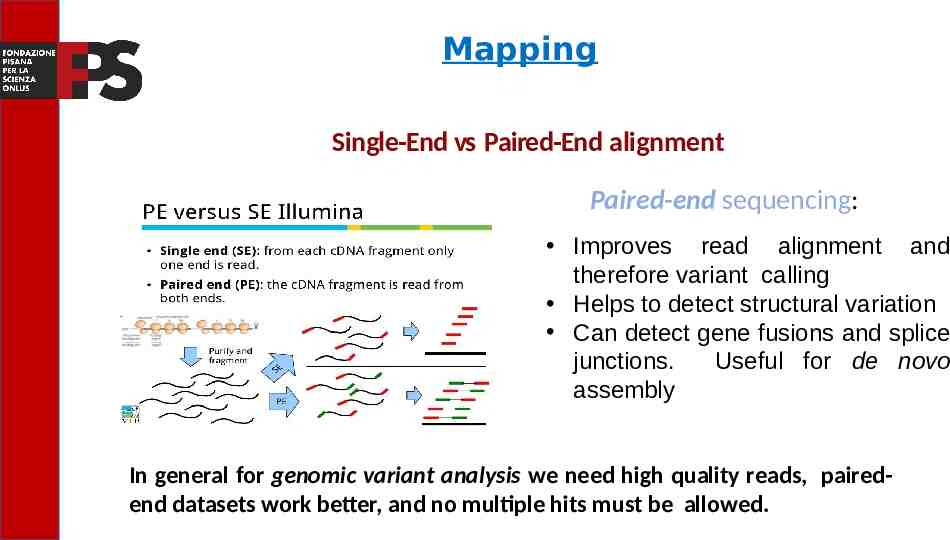
Mapping Single-End vs Paired-End alignment Paired-end sequencing: Improves read alignment and therefore variant calling Helps to detect structural variation Can detect gene fusions and splice junctions. Useful for de novo assembly In general for genomic variant analysis we need high quality reads, pairedend datasets work better, and no multiple hits must be allowed.
Mapping Before starting the mapping get a reference genome! A reference genome is a consensus sequence built up from high quality sequenced samples from different populations. It is the control reference sequence to compare our samples Genome Reference Consortium (GRC) created to deliver assemblies: http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/ Current human assembly is GRCh38, released in the summer of 2014. Many genomic regions corrected and improved such as centromeres.
FASTA Files The file that stores the data of the reference genomes is in the fasta format. It is a text format that has a first line of header, where there are data related to the IDs of the chromosomes
Sequence Mapping Difficulties: The high volume of data and the size of the reference genome constitute one of the major difficulties from the computational point of view, reflecting on the execution times. The length of the reads and the ambiguity caused by repeats and sequence errors are reflected in the accuracy of the mapping.
How to choose an aligner? There are many short read aligners and they vary a lot in performance (accuracy, memory usage, speed and flexibility etc). Factors to consider : application, platform, read length, downstream analysis, etc. Guaranteed high accuracy will take longer time. Popular choices: Bowtie2, BWA, Tophat2, STAR.
Mapping: RNA-seq TopHat2, the (old) standard RNA-seq aligner: It uses Bowtie2 to align reads, so it is not very sensitive, usually maps 75% of reads. Not ready for long reads ( 150bp), mapping decrease to below 50%. Poor performance, can take several hours to map. Mapping fall down with mismatches, INDELS and longer reads. STAR: STAR developed for ENCODE project High-performance, not very high sensitivity.
Mapping: DNA-seq BWA: It was one of the first NGS mappers and is the most widely used, provides very good results in common scenarios (genome and exome analysis). It is multi-thread, but lacks some features such as support for RNA-seq or big INDELS (Insertions – Deletions). Not specially fast. Bowtie2 Bowtie2 is claimed to be the fastest, but it missed many reads. It is a little bit less sensitivity than BWA. Fail to correctly map many mismatches and INDELS. It is multi-thread, but lacks some biological features such as support for RNA or big INDELS.
Outline Introduction Bioinformatics in NGS data analysis 1. 2. 3. 4. Basics: terminology, data formats, general workflow etc Data Analysis Pipeline Sequence QC and preprocessing Sequence mapping DNA-Seq data analysis 1. Post-alignment processing 2. Variant Calling 3. Variant Annotation, Filtration, Prioritization 4. NGS and rare diseases Summary
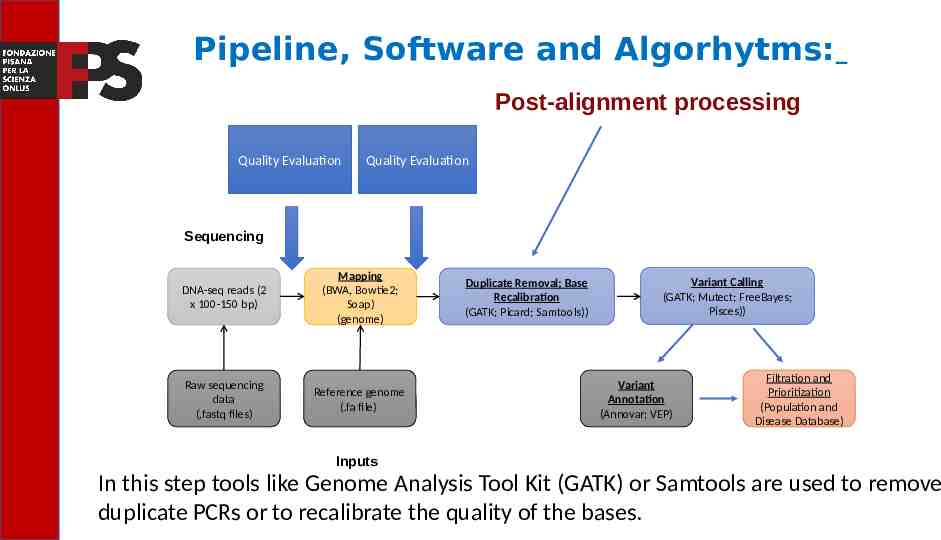
Pipeline, Software and Algorhytms: Post-alignment processing Quality Evaluation Quality Evaluation Sequencing Read alignment DNA-seq reads (2 x 100-150 bp) Mapping (BWA, Bowtie2; Soap) (genome) Raw sequencing data (.fastq files) Reference genome (.fa file) Transcript compilation Duplicate Removal; Base Recalibration (GATK; Picard; Samtools)) Differential expression Variant Calling (GATK; Mutect; FreeBayes; Pisces)) Variant Annotation (Annovar; VEP) Filtration and Prioritization (Population and Disease Database) Inputs In this step tools like Genome Analysis Tool Kit (GATK) or Samtools are used to remove duplicate PCRs or to recalibrate the quality of the bases.
Duplicates Removal Creating duplicate PCRs during sample preparation can lead to problems in the detection of genetic variants. . A sequencing error could be propagated by generating a genetic variant that is really a false positive. We can remove the PCR duplcate by using bioinformatics tools (GATK, Picard, Samtools.
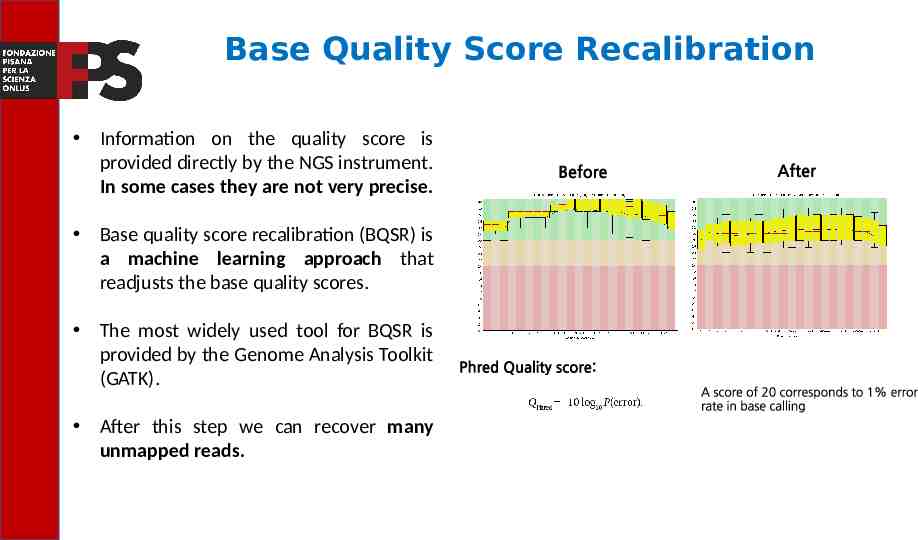
Base Quality Score Recalibration Information on the quality score is provided directly by the NGS instrument. In some cases they are not very precise. Base quality score recalibration (BQSR) is a machine learning approach that readjusts the base quality scores. The most widely used tool for BQSR is provided by the Genome Analysis Toolkit (GATK). After this step we can recover many unmapped reads.
Outline Introduction Bioinformatics in NGS data analysis 1. 2. 3. 4. Basics: terminology, data formats, general workflow etc Data Analysis Pipeline Sequence QC and preprocessing Sequence mapping DNA-Seq data analysis 1. Post-alignment processing 2. Variant Calling 3. Variant Annotation, Filtration, Prioritization 4. NGS and rare diseases Summary Following data processing steps, the reads are ready for downstream analyses. In the case of DNA-seq analysis the following step is most frequently Variant Calling.
Variant Calling Variant calling is the process of identifying differences between the sequencing reads and a reference genome. Input file: BAM-file Output file: Variant Caller Format - file (VCF)
Variant Calling: VCF file Variant Caller Format file (VCF) is a very raw output of the variant calling process. It contains the chromosomal coordinates of the mutations, useful information to extrapolate the type of mutation, the name of the sample etc. No gene information inside
Variant Calling: VCF file Another very important information in the VCF files is the one related to the Variant Allele Frequency (VAF). This value indicates how many reads support the presence of genetic variation. 12% 21%
Variant Calling The most widely used state-of-the-art variant callers include, GATKHaplotypeCaller, SOAPsnp, SAMTools, bcftools, Strelka, FreeBayes, Platypus, and DeepVariant. A combination of different variant callers outperforms any single method
Somatic calling – some tools To detect mutations in cancer samples there are specific tools: MuTect2, VarScan2, SomaticSniper. Almost all of them require the presence of normal tissue data matched with tumor tissue from the same individual to highlight the presence of genetic alterations present only in the tumor sample. The only tool able to operate with only the presence of tumor data is Pisces (from Illumina), which infers somatic mutations on the basis of the low Variant Allele Frequency.
Outline Introduction Bioinformatics in NGS data analysis 1. 2. 3. 4. Basics: terminology, data formats, general workflow etc Data Analysis Pipeline Sequence QC and preprocessing Sequence mapping DNA-Seq data analysis 1. Post-alignment processing 2. Variant Calling 3. Variant Annotation, Filtration, Prioritization 4. NGS and rare diseases Summary
Variant Annotation, Filtration, Prioritization Next-generation sequencing generates thousands of sequence variants that must be filtered and prioritized for clinical interpretation
Variant Annotation, Filtration, Prioritization This process may differ slightly among individual laboratories, but it generally includes annotation of variants (mainly to attribute the variant to a specific gene or transcript), application of population frequency filters and database searches to enrich for rare variants and eliminate common variants, and prediction of functional effect.
VARIANT ANNOTATION Variant annotation is a critical step in the genomic analysis workflow. The aim of all functional annotation tools is to annotate information of the variant effects/consequences, including: 1. Listing which genes/transcripts are affected. 2. Determination of the consequence on protein sequence. 3. Correlation of the variant with known genomic annotations (e.g., coding sequence, intronic sequence, noncoding RNA, regulatory regions, etc.). 4. matching known variants found in variant databases (dbSNP , 1000 Genomes Project, ExAc, gnomAD, COSMIC, ClinVar)
VARIANT ANNOTATION Once the analysis-ready VCF is produced, the genomic variants can then be annotated using a variety of tools and a variety of transcript sets. Both the choice of annotation software and transcript set (e.g., RefSeq transcript set, Ensembl transcript set) have been shown to be important for variant annotation. The most widely used functional annotation tools include: AnnoVar, SnpEff, Variant Effect Predictor (VEP), GEMINI , VarAFT VAAST, TransVar, MAGI, SNPnexus, and VarMatch.
VARIANT ANNOTATION Many annotation tools utilize the predictions of SNV/indel pathogenecity prediction methods, to name a few, SIFT, PolyPhen-2, LRT, MutationTaster, MutationAssessor, FATHMM, GERP , PhyloP, SiPhy , PANTHER-PSEP [43], CONDEL, CADD, CHASM, CanDrA, and VEST.
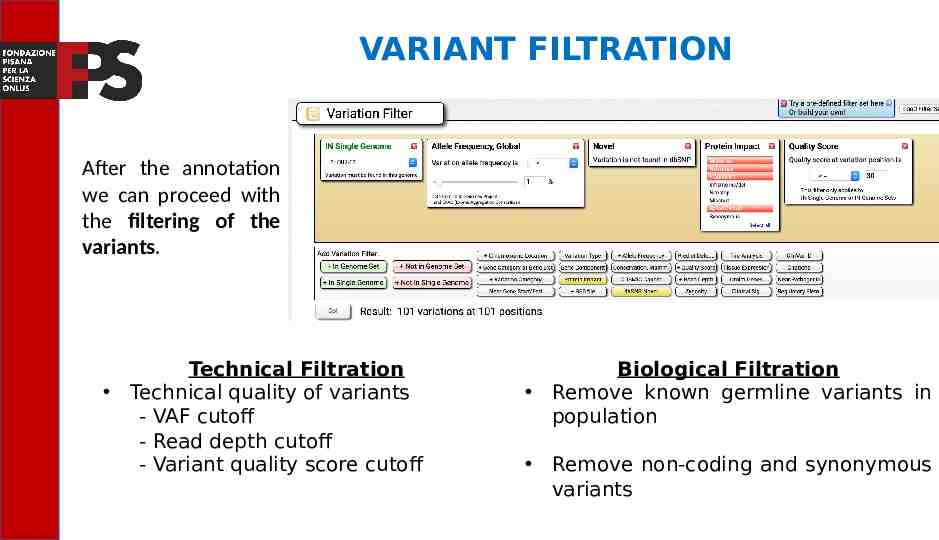
VARIANT FILTRATION After the annotation we can proceed with the filtering of the variants. Technical Filtration Technical quality of variants - VAF cutoff - Read depth cutoff - Variant quality score cutoff Biological Filtration Remove known germline variants in population Remove non-coding and synonymous variants
VARIANT PRIORITIZATION The most difficult aspect is to give biological and clinical meaning to the impressive number of genetic variants detected through WES/WGS.
VARIANT PRIORITIZATION Methods required for the interpretation of genomic variants: variant-dependent annotation such as population allele frequency (e.g., in 1000 Genomes, ExAc, gnomAD); the predicted effect on protein and evolutionary conservation; disease-dependent inquiries such as mode of inheritance; co-segregation of variant with disease within families; prior association of the variant/gene with disease, investigation of clinical actionability; pathway-based analysis;
VARIANT PRIORITIZATION Mutational databases are an indispensable resource to give meaning to genetic data. We can verify if, for example, a mutation has already been found and associated with a disease. Databases such as ClinVar, HGV databases, COSMIC, and CIViC can aid interpretations of clinical significance of germline and somatic variants for reported conditions.
VARIANT PRIORITIZATION Some software helps to speeds up the process of interpreting variants. Ingenuity Variant Analysis, BaseSpace Variant Interpreter, VariantStudio, Varaft and Phenoxome
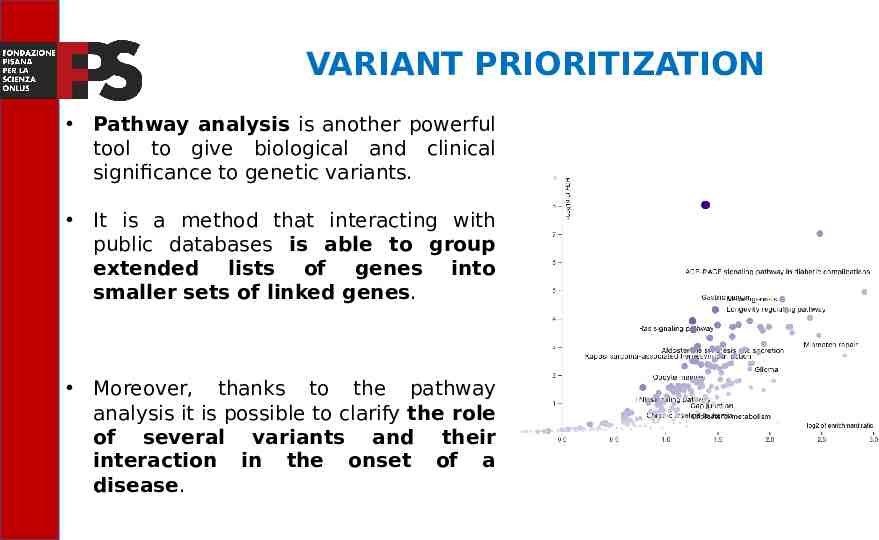
VARIANT PRIORITIZATION Pathway analysis is another powerful tool to give biological and clinical significance to genetic variants. It is a method that interacting with public databases is able to group extended lists of genes into smaller sets of linked genes. Moreover, thanks to the pathway analysis it is possible to clarify the role of several variants and their interaction in the onset of a disease.
VARIANT PRIORITIZATION Countless tools for pathway analysis exist. Some of the widely used pathway analysis tools are GSEA, DAVID, IPA PathVisio. Additionally, many different pathway resources exist, the most popular of which are Kyoto Encyclopedia of Genes and Genomes (KEGG), Reactome, WikiPathways, MSigDb, STRINGDB, Pathway Commons, Ingenuity Knowledge Base, and Pathway Studio. WEB-based Gene SeT AnaLysis Toolkit (WEBGESTALT) brings together methods and databases to perform a very comprehensive analysis.
VARIANT PRIORITIZATION Many times, however, it is necessary to validate «in vitro» the results in silico, in order to arrive at definitive conclusions about the pathogenicity of genetic alterations. Especially if you want to inform a patient of the course of the disease. Functional validation can be performed using different model systems (e.g., patient cells, model cell lines, model organisms, induced pluripotent stem cells) and performing the suitable type of assay (e.g., genetic rescue, overexpression, biomarker analysis).
Outline Introduction Bioinformatics in NGS data analysis 1. 2. 3. 4. Basics: terminology, data formats, general workflow etc Data Analysis Pipeline Sequence QC and preprocessing Sequence mapping DNA-Seq data analysis 1. Post-alignment processing 2. Variant Calling 3. Variant Annotation, Filtration, Prioritization 4. NGS and rare diseases Summary
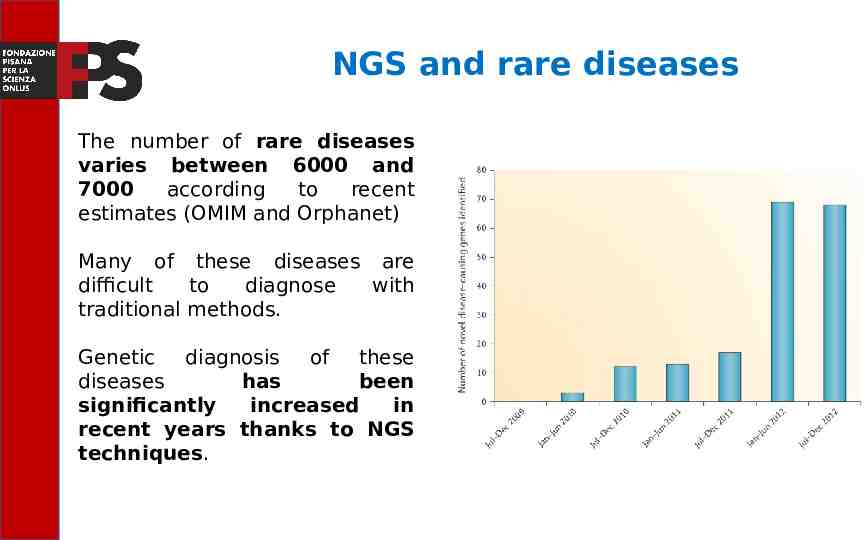
NGS and rare diseases The number of rare diseases varies between 6000 and 7000 according to recent estimates (OMIM and Orphanet) Many of these diseases are difficult to diagnose with traditional methods. Genetic diagnosis of these diseases has been significantly increased in recent years thanks to NGS techniques.
NGS and rare diseases WGS and WES are powerful approaches for detecting genetic variation. However, because of the extent and inherent complexity (as well as the greater cost) of WGS, WES is currently the more popular platform for the discovery of rare-disease-causing genes. A clinical next-generation sequencing test can be designed to target a panel of selected genes. Gene panels target curated sets of genes associated with specific clinical phenotypes.
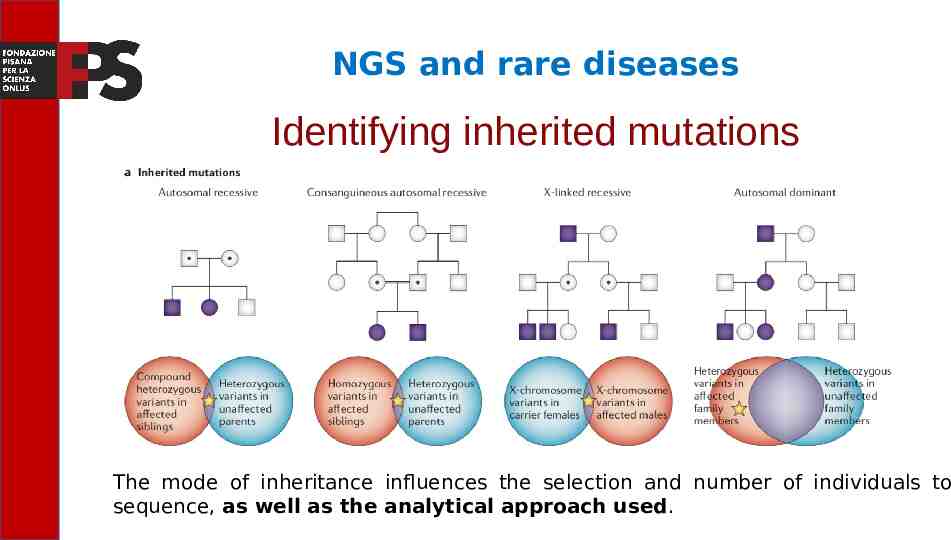
NGS and rare diseases Identifying inherited mutations Knowledge of the disease history of the various family members is always of great help in the diagnosis of genetic diseases When there is familial recurrence of a defined rare phenotype or parental consanguinity, the likelihood that a rare disease is monogenic is high.
NGS and rare diseases Identifying inherited mutations The mode of inheritance influences the selection and number of individuals to sequence, as well as the analytical approach used.
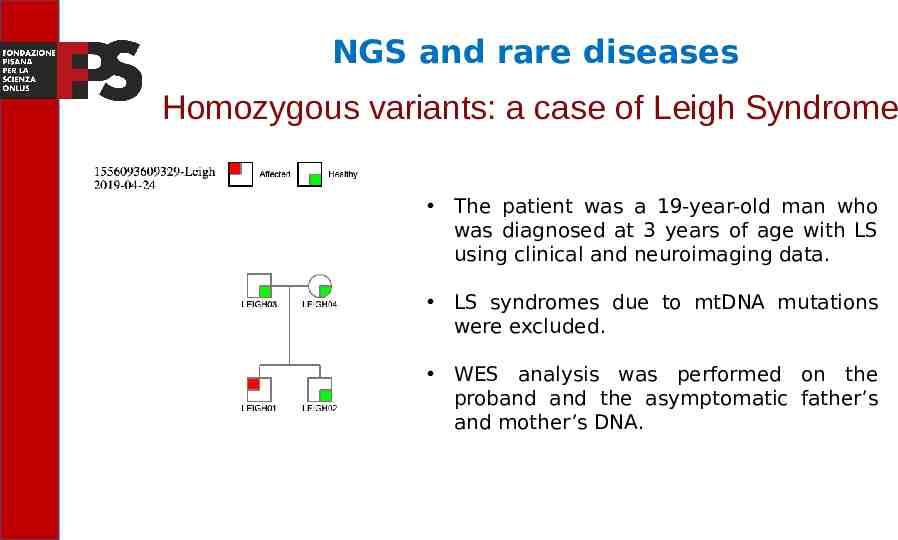
NGS and rare diseases Autosomal recessive disease: a case of Leigh Syndrome We have supported a unit of Medical Genetics to correctly frame a case of Leigh Syndrome, a neurodegenerative disease that leads to death in the early years of life. The disease is generally caused by mutations in mitochondrial genes, although mutations also exist in nuclear genes. We decided to approach the case by analyzing the members of the family with the WES.
NGS and rare diseases Homozygous variants: a case of Leigh Syndrome The patient was a 19-year-old man who was diagnosed at 3 years of age with LS using clinical and neuroimaging data. LS syndromes due to mtDNA mutations were excluded. WES analysis was performed on the proband and the asymptomatic father’s and mother’s DNA.
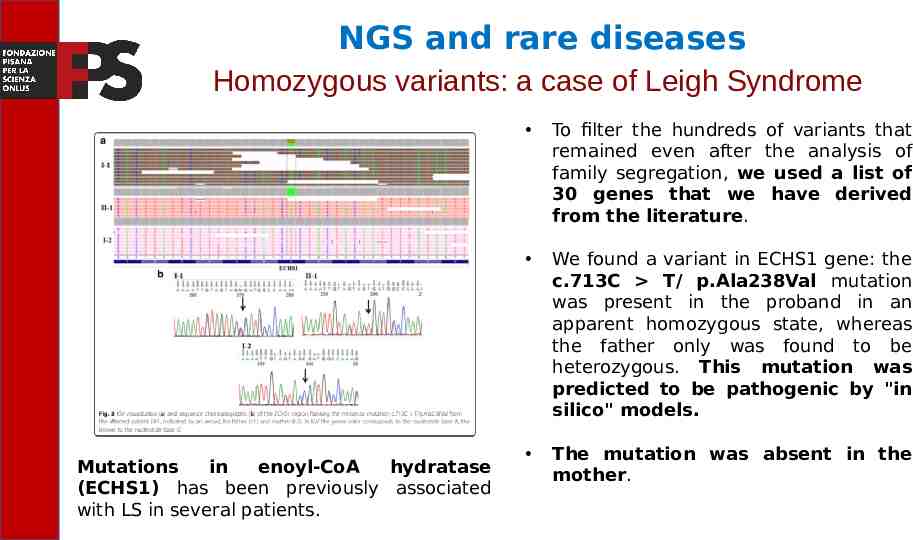
NGS and rare diseases Homozygous variants: a case of Leigh Syndrome Mutations in enoyl-CoA hydratase (ECHS1) has been previously associated with LS in several patients. To filter the hundreds of variants that remained even after the analysis of family segregation, we used a list of 30 genes that we have derived from the literature. We found a variant in ECHS1 gene: the c.713C T/ p.Ala238Val mutation was present in the proband in an apparent homozygous state, whereas the father only was found to be heterozygous. This mutation was predicted to be pathogenic by "in silico" models. The mutation was absent in the mother.
NGS and rare diseases Homozygous variants: a case of Leigh Syndrome Generally a homozygous mutation exists because both mother and father carry the same mutation. This made us suspect the presence of a deletion of one portion of chromosome where the ECHS1 gene is located. We detected a deletion in an extended region of chromosome 10 (from 135,120,573 to 135,187,238) involving five genes: ZNF511, CALY, PRAP1, FUOM, and ECHS1. This deletion was present in the proband and in his mother but not in the father.
NGS and rare diseases Homozygous variants: a case of Leigh Syndrome We confirmed the clinical diagnosis hypothesized for 15 years by using whole exome sequencing (WES) analysis, which identified a missense mutation in ECHS1 and a deletion of the entire gene.
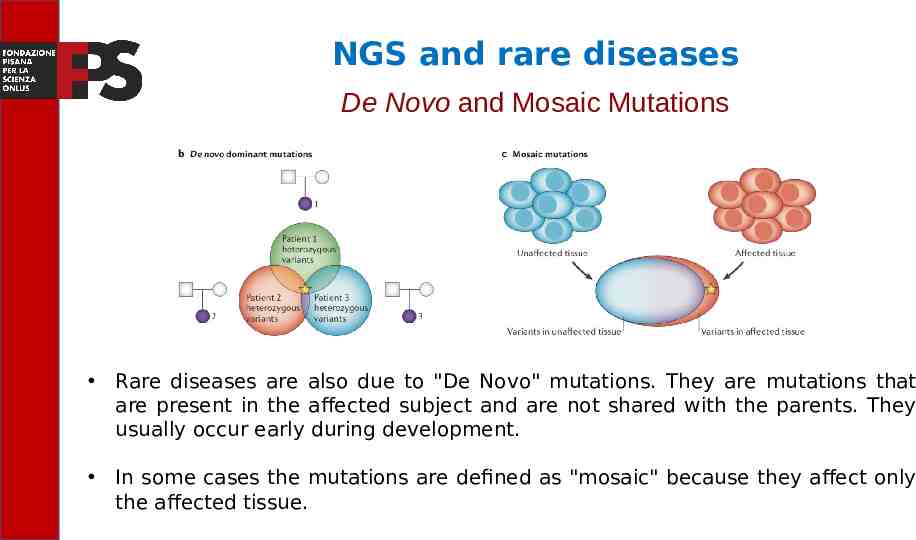
NGS and rare diseases De Novo and Mosaic Mutations Rare diseases are also due to "De Novo" mutations. They are mutations that are present in the affected subject and are not shared with the parents. They usually occur early during development. In some cases the mutations are defined as "mosaic" because they affect only the affected tissue.
NGS and rare diseases Autosomal dominant disorders: de novo mutations De novo mutations causing autosomal dominant disorders have proved to be much easier to identify, given that each individual carries very few variants that are not also found in their parents, resulting in a data set that is much less complex.
NGS and rare diseases Autosomal dominant disorders: de novo mutations Prematurely deceased child with arthrogryposis (congenital joint contracture in two or more areas of the body.), hypotonia, urinary problems and neurodevlopmental delay. Initially diagnosed as suffering from Congenital Multiplex Arthrogryposis. Negative to genetic investigation for known causative genes
NGS and rare diseases Autosomal dominant disorders: de novo mutations After WES analysis, we analyzed the data using Phenoxome, a web tool, which annotates the genetic variants associating them to phenotypic manifestations of the disease. Phenoxome adopt a robust phenotype-driven model to facilitate automated variant prioritization. Phenoxome dissects the phenotypic manifestation of a patient in concert with their genomic profile to filter and then prioritize variants that are likely to affect the function of the gene (potentially pathogenic variants).
NGS and rare diseases Autosomal dominant disorders: de novo mutations Phenoxome returned a mutation of the PURA gene, as a likely mutation candidate to explain the patient's symptoms. This mutation is a nonsense mutation, which therefore leads to a premature end of the protein, dramatically altering its structure. This mutation is present only in the affected subject
NGS and rare diseases Autosomal dominant disorders: de novo mutations PURA encodes Pur-α, a highly conserved multifunctional protein that has an important role in normal postnatal brain development in animal models. Mutations in the PURA gene have recently been associated with the symptoms described in our patient.
SUMMARY The advancements in NGS and the development of bioinformatics methods and resources enabled the usage of WES/WGS to detect, interpret, and validate genomic variations in the clinical setting. As we attempted to describe WES/WGS analysis is challenging, and there are a great number of tools for each step of variation discovery. An optimal and coordinated combination of tools is required to identify the different types of genomic variants. Patients with rare genetic diseases are among the first beneficiaries of the NGS revolution; their experience will inform personalized medicine in other areas over the next decade.






