School of Pharmacy & Pharmaceutical Sciences Annex 1: Changes






























31 Slides3.86 MB
School of Pharmacy & Pharmaceutical Sciences Annex 1: Changes & Interpretations Declan Kelly Head of Quality, Sanofi Ireland QP Forum 2018 – Thursday 12th April
Overview Background Impact on the QP Changes in Annex 1 Topics for discussion Group Review QP Forum 2018 – Thursday 12th April
Revised Annex 1: Background In December 20, 2017, the long anticipated draft of updated Annex 1 was published (Manufacture of Sterile Medicinal Products). Annex 1 was also previously partially revised in 1996, 2003 and 2007. Manufacturing Technologies and GMP have both advanced considerably since the previous revision – including ICH Q9 and Q10. Current draft revision was intended as a complete re-write of the Annex by a combined working group of EMA, PIC/S and also WHO. Current Annex 1 consists of 16 pages, the new document with its additions and changes now amounts to 50 pages. One interesting aspect of the latest revision is that the revised Annex 1 is not just an independent EU document but also applies directly to the PIC/S guidelines. As a result, the planned revision replaces the current versions of EU-GMP Annex 1 and also the PIC/S document PE 009 -11 "Manufacture of sterile Medicinal Products". The new ‘Scope’ & ‘Principles’ provide valuable regulatory insight. QP Forum 2018 – Thursday 12th April
Revised Annex 1: Impact on the QP So, what requirements are specifically listed for the QP? Due to the alignment with PIC/S the draft does not actually contain any direct references to the term "Qualified Person" (QP); instead the QP role is referenced in the document as the "Person responsible for the quality release of sterile medicines". There are a number of points to consider for QPs: The ‘responsible person’ should have "appropriate access to manufacturing and quality information and possess adequate knowledge and experience in the manufacture of sterile dosage forms and their critical quality attributes." However a number of items are listed specifically that need to be considered in the batch certification process: Where used for aseptic manufacturing, confirmation of the integrity of the final sterilization gas filter should be considered as part of the batch release process [7.19]. Sterilization records should be available for each sterilisation run. These should be reviewed and approved as part of the batch release procedure [8.40]. QP Forum 2018 – Thursday 12th April
Revised Annex 1: Impact on the QP Contamination Control Strategy (Batch Release aspects): In Annex 1, the EM and process monitoring program should be part of an overall contamination control strategy (CCS). The goal is to minimise the risk of both microbial and particulate contamination. The information from the CCS systems should be "used for routine batch release and for periodic assessment during process review or investigations" [9.3]. Results from monitoring surfaces and personnel and environmental monitoring data generated in grade A and B should also be considered during batch record review [9.11], [10.10]. Where Parametric Release is performed the following should also be considered: A robust system should be applied to the product lifecycle validation and the routine monitoring of the manufacturing process. This system should be periodically reviewed [8.48]. If a sterilization with ethylene oxide is performed (which should only be used when no other method is practicable), each sterilization cycle should be monitored with suitable biological indicators, using the appropriate number of test pieces distributed throughout the load unless parametric release has been authorized by the National Competent Authority [8.75].
Changes in Annex 1 from 2008 version In expanding from 16 to 50 pages there are multiple new and extended areas in the draft document. In this section we will cover a selection of those changes (plus some notable items that have not changed) in draft Annex 1 QP Forum 2018 – Thursday 12th April
Annex 1 - changes Section 1.0 Scope - Introduction of principles of QRM: This Annex provides general guidance that should be used for all sterile medicinal products and sterile active substances, via adaption, using the principles of Quality Risk Management (QRM), to ensure that microbial, particulate and pyrogen contamination associated with microbes is prevented in the final product. QP Forum 2018 – Thursday 12th April
Annex 1 - changes Section 2.0 Principle – Manufacture of Sterile Products: a) Facility, equipment and process design must be optimized qualified and validated according to Annex 11 and Annex 15 of EU GMP. The use of appropriate current technologies should be implemented to ensure protection and control of the product from potential extraneous sources of particulate and microbial contamination such as personnel, materials and the surrounding environment. b) Personnel must have appropriate skills, training and attitudes with a specific focus on the principles involved in the protection of sterile product during the manufacturing, packaging and distribution processes.
Annex 1 - changes Section 2.0 Principle – Introduction of Contamination Control Strategy: Quality Assurance is particularly important, and manufacture of sterile products must strictly follow carefully established and validated methods of manufacture and control. A contamination control strategy should be implemented across the facility in order to assess the effectiveness of all the control and monitoring measures employed. This assessment should lead to corrective and preventative actions being taken as necessary. The strategy should consider all aspects of contamination control and its life cycle with ongoing and periodic review and update of the strategy as appropriate.
Annex 1 - changes Section 2.0 Principle – Introduction of Contamination Control Strategy: Elements to be considered within such a documented contamination control strategy should include (but not be limited to): a) Design of both the plant and process. b) Equipment and facilities. c) Personnel. d) Utilities. e) Raw Materials Control – including in-process controls. f) Product containers and closures. g) Vendor approval – such as key component suppliers, sterilization of components and single use systems, and services. h) For outsourced services, such as sterilization, sufficient evidence should be provided 86 to the contract giver to ensure the process is operating correctly. i) Process risk assessment. j) Process validation. k) Preventative maintenance – maintaining equipment and premises (planned and unplanned maintenance) to a standard that will not add significant risk of contamination. l) Cleaning and disinfection. m) Monitoring systems - including an assessment of the feasibility of the introduction of scientifically sound, modern methods that optimize the detection of environmental contamination. n) Prevention – Trending, investigations, corrective and preventive actions (CAPA), root cause determination and the need for more robust investigational tools. o) Continuous improvement based on information from the above systems
Annex 1 - changes Section 5.0 Premises: 5.20 For isolators, the required background environment can vary depending on the design of the isolator, its application and the methods used to achieve bio-decontamination*. The decision as to the supporting background environment should be documented in a risk assessment where additional risks are identified, such as for negative pressure isolators. Where items are introduced to the isolator after disinfection then a higher grade of background should be considered *Note: ‘Bio-decontamination’ not defined
Annex 1 - changes Section 6.0 Equipment: 6.3 . if maintenance has to be performed in the clean area then precautions such as additional disinfection and additional environmental monitoring should be considered. If sterilization is required, it should be carried out, wherever possible, after complete reassembly. QP Forum 2018 – Thursday 12th April
Annex 1 - changes There is a new chapter on "Utilities", meaning the required equipment and/or other materials that may come into contact with a product or influence it directly. The section covers water systems, steam used for sterilization, compressed gas and vacuum and cooling systems. 7.0 Utilities: 7.2 In general higher risk utilities are those that: a) Directly contact product e.g. compressed gases. b) Contact materials that ultimately will become part of the product. c) Control contamination of surfaces that contact the product. d) Or otherwise directly impact the product 7.4 Results for critical parameters of the high risk utility should be subject to regular trend analysis to ensure that system capabilities remain appropriate QP Forum 2018 – Thursday 12th April
Annex 1 - changes 7.0 Utilities: Water Systems 7.12 Where WFI storage tanks are equipped with hydrophobic bacteria retentive vent filters the filters should be sterilized, and the integrity of the filter tested before and after use. 7.15 Regular ongoing chemical and microbial monitoring of water systems should be performed with alert limits based on the qualification that will identify an adverse trend in the performance of the systems. Sampling should include all outlets and user points at a specified interval. A sample from the worst case sample point, e.g. the end of the distribution loop return, should be included each time the water is used for manufacturing and manufacturing processes. QP Forum 2018 – Thursday 12th April
Annex 1 - changes Section 8.0 Production & Specific Technologies: Aseptic Preparation Examples of operations and which grades they should be performed in Current Annex 1: Annex 1 Revision:
Annex 1 - changes Finishing of sterile products: Visual Inspection 8.26 Batches with unusual levels of defects from visual inspection , when compared to routine defect levels for the process, should lead to investigation and consideration of partial or the whole rejection of the batch concerned . Critical defects should not be identified during any subsequent sampling of acceptable containers as it indicates a failure of the original inspection process. Q: It is known that Visual Inspection is never 100% effective – does this imply you can never re-inspect a batch? It may also cause a problem for companies supplying Japan where local inspections are typically routinely performed. If the Japanese inspectors find any critical defects it indicates a failure of the original insection.
Annex 1 – “The Old Reliables, still here” Finishing of sterile products: ‘CCI’ 8.18 Containers should be closed by appropriately validated methods. Containers closed by fusion, e.g. Form-Fill-Seal Small Volume Parenteral (SVP) & Large Volume Parenteral (LVP) bags, glass or plastic ampoules, should be subject to 100% integrity testing. Samples of other containers should be checked for integrity utilising validated methods and in accordance with QRM, the frequency of testing should be based on the knowledge and experience of the container and closure systems being used. A statistically valid sampling plan should be utilized. It should be noted that visual inspection alone is not considered as an acceptable integrity test method Filtration of medicinal products which cannot be sterilized in their final container: ‘PUPSIT’ 8.84 The integrity of the sterilized filter assembly should be verified by testing before use, in case of damage and loss of integrity caused by processing, and should be verified by on line testing immediately after use by an appropriate method such as a bubble point, diffusive flow, water intrusion or pressure hold test. QP Forum 2018 – Thursday 12th April
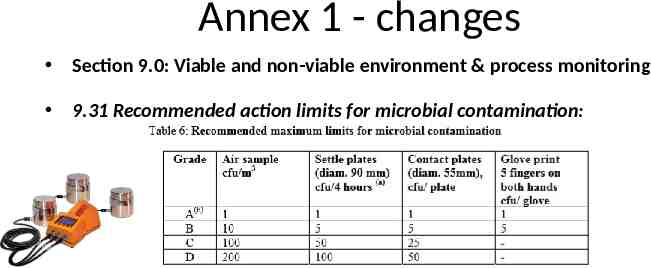
Annex 1 - changes Section 9.0: Viable and non-viable environment & process monitoring 9.31 Recommended action limits for microbial contamination: 2008 (Current version) Annex 1: Note currently 1 cfu
Annex 1 - changes 9.4 Environmental Monitoring 9.4 In order to establish a robust environmental monitoring program, i.e. locations, frequency of monitoring and incubation conditions (e.g. time, temperature(s) and aerobic and or anaerobic), appropriate risk assessments should be conducted based on detailed knowledge of the process inputs, the facility, equipment, specific processes, operations involved and knowledge of the typical microbial flora found, consideration of other aspects such as air visualization studies should also be included. These risk assessments should be re-evaluated at defined intervals in order to confirm the effectiveness of the site’s environmental monitoring program, and they should be considered in the overall context of the trend analysis and the contamination control strategy for the site. QP Forum 2018 – Thursday 12th April
Annex 1 - changes Environmental Monitoring (continued) 9.8 Appropriate alert and action limits should be set for the results of particulate and microbiological monitoring. Alert levels should be established based on results of Performance Qualification (PQ) tests or trend data and should be subject to periodic review. 9.9 The alert limits for grade B, C and D should be set based on the area performance, with the aim to have limits lower than those specified as action limits, in order to minimise risks associated and identify potential changes that may be detrimental to the process QP Forum 2018 – Thursday 12th April
Annex 1 - changes 9.13 The recommended limits for airborne particle concentration in monitoring for each grade are given in Table 5 (below). New New QP Forum 2018 – Thursday 12th April
Annex 1 - changes Viable Monitoring 9.28 Rapid microbial monitoring methods may be adopted after validation as long as they are demonstrated to be at least equivalent to the established methodology. 9.32 Monitoring procedures should define the approach to trending. Trends can include but are not limited to: a) Increasing numbers of action or alert limit breaches. b) Consecutive breaches or alert limits. c) Regular but isolated breaches of limits that may have a common cause, for example single excursions that always follow planned preventative maintenance. d) Changes in flora type and numbers. QP Forum 2018 – Thursday 12th April
Annex 1 - changes Viable Monitoring 9.33 If microorganisms are detected in a grade A or B zone, they should be identified to species level and the impact of such microorganisms on product quality (for each batch implicated) and state of control should be evaluated. Consideration may also be given to the identification of grade C and D contaminants and the requirements should be defined in the contamination control strategy. QP Forum 2018 – Thursday 12th April
Annex 1 - changes Aseptic process simulation (“media fills”): 9.35 f) The process simulation test for lyophilized products should include the entire aseptic processing chain, including filling, transport, loading, chamber dwell, unloading and sealing. The process simulation should duplicate the lyophilization process, with the exception of freezing and sublimation, including partial vacuum and cycle duration and parameters as appropriate for the media. Boiling over or actual freezing of the solution should be avoided 9.36 The process simulation testing should take into account various aseptic manipulations and interventions known to occur during normal production as well as worst-case situations, including: a) Inherent interventions at the maximum accepted frequency per number of filled units. b) Corrective interventions in representative number and with the highest degree of intrusion acceptable. 9.38 In developing the process simulation test plan, risk management principles should be used
Annex 1 - changes Aseptic process simulation (“media fills”): 9.40 Process simulation tests should be performed as initial validation, generally with three consecutive satisfactory simulation tests per shift, and after any significant modification to the HVAC system, equipment, major facility shut down, process and number of shifts, etc. Normally process simulation tests (periodic revalidation) should be repeated twice a year (approximately every six months) for each aseptic process and filling line, and at least annually for each operator. 9.43 The target should be zero growth and any contaminated unit should result in an investigation to determine the root cause (if possible) and to identify appropriate CAPA. Following implementation of CAPA, a repeat APS will be required to validate the effectiveness of the CAPA. The number of APS to be repeated should be determined using QRM principles taking into consideration the number and type of CAPA and the level of contamination found in the failed APS. Typically 3 successful consecutive repeat APS would be expected; any differences to this expectation should be clearly justified prior to repeat performance
Annex 1 – Other Discussion Topics 11. Glossary: Note the Glossary also contains additional guidance, not just definitions of terms. Could be audited against this. 8.30 Where possible, finished product should be terminally sterilized using a validated sterilization process as this provides a greater assurance of sterility than a validated and controlled sterilizing filtration process and/or aseptic processing. Where it is not possible for a product to undergo a sterilisation, consideration should be given to using terminal bioburden reduction steps, such as heat treatments (pasteurization), combined with aseptic processing to give improved sterility assurance. 8.47 Moist heat sterilization: No mention of steam quality testing is included 4.13 Outdoor clothing should not be brought into changing rooms leading to Grade B and C rooms. It is recommended that facility suits, including dedicated socks, be worn before entry to change rooms for grade C and B. Where clothing is reused this should be considered as part of the qualification. Q: More guidance needed on sterile gloves/scrubs too? Q: Is there a higher risk removing socks and shedding particles? QP Forum 2018 – Thursday 12th April
Annex 1 – Other Discussion Topics Aseptic process simulation (“media fills”): while it provides more detail than the current version, does the revised draft guidance provide enough clarity on APS? 5.34 Fumigation or vapour disinfection of clean areas such as Vapour Hydrogen Peroxide (VHP) may be useful for reducing microbiological contamination in inaccessible places. There is a lack of guidance in draft Annex 1 for widely used VHP – which may leave it open to interpretation. For example, should expectations on pre-sterilised stopper bowls, which are then assembled in isolators, followed by a VHP cycle, be included? Guidance on management of glove issues for Isolators/RABS would be useful. QP Forum 2018 – Thursday 12th April
Annex 1 – Other Discussion Topics 4.16 The ambient temperature and humidity should be set to prevent shedding due to operators becoming too cold (leading to excessive movement) or too hot. – Q: humidity controls may impact older facilities. May need retrofitting? 9.35 (f): APS section: Duplicating a lyo cycle including cycle duration (up to 5 days in some cases) for APS may be challenging. Could a risk assessment including chamber leak testing & filter integrity etc. support justifying a reduced simulated cycle duration? General comment: the word ‘Integrity’ is used 43 times in the draft Annex 1 (for units, filters, transport mechanisms etc). What is actually required for integrity is never specified or defined. QP Forum 2018 – Thursday 12th April
Annex 1 – Other Discussion Topics 8.84: For PUPSIT the use of a redundant second 0.2um filter is not discussed, nor how this should be managed in terms of the draft guidance, e.g. taking bioburden samples before the first filter & the sterilising filter etc. – (Note: Section 8.78 does refer to use of a second sterilising grade filter: Due to the potential additional risks of a sterilizing filtration process as compared to other sterilization processes, a second filtration through a sterile, sterilising grade filter (positioned as per clause 8.15), immediately prior to filling, is advisable). 8.84 continued: The approach for small batch sizes: It is recognised that for small batch sizes, this (PUPSIT) may not be possible; in these cases an alternative approach may be taken as long as a formal risk assessment has been performed and compliance is achieved. – Q: how small is a small batch size? Is it intended for IMPs only? QP Forum 2018 – Thursday 12th April
Annex 1 – Other Discussion Topics ‘Sanitisation’ has been replaced by ‘Cleaning & Disinfection’ in the draft. This is a positive development bringing additional clarity for industry. Q: The introduction of QRM into the document is a very positive one but is it contradicted by some of the very prescriptive text? QP Forum 2018 – Thursday 12th April
Thank You Questions? Comments? Feedback?






