Regulatory Binder: Best Practices Derita Bran MSN, RN, CCRC Program


























































59 Slides2.38 MB
Regulatory Binder: Best Practices Derita Bran MSN, RN, CCRC Program Director Tennessee Clinical and Translational Science Institute (TN-CTSI)
Objectives By the end of this module, the participant will be able to: Describe the purpose of the Regulatory Binder Discuss best practices when maintaining a Regulatory Binder Identify essential documents maintained within the Regulatory Binder
Disclaimer THE INFORMATION PROVIDED IS AN EXAMPLE OF THE REGULATORY BINDER SET-UP AND THE CONTENTS TO BE MAINTAINED SECTIONS DISCUSSED MAY NOT APPLY TO ALL STUDIES THIS SLIDE PRESENTATION IS OF GENERAL INFORMATION AND SHOULD NOT BE CONSIDERED ALL-INCLUSIVE OF INFORMATION REGARDING THE REGULATORY BINDER THE PRESENTER HAS NO FINANCIAL OR OTHER INTEREST RELEVANT TO THE SUBJECT MATTER *THIS PRESENTATION IS SPONSORED BY TN-CTSI
What is a Regulatory Binder? Electronic/paper/combination file that contains: Study specific information Study essential documents Regulatory documents
Regulatory Binder (RB) Purpose: Compliance: Demonstrates compliance with Regulations and Good Clinical Practice (GCP) Validates adherence to high standards of research practices and human subject protection Record Keeping: Provides an organizational framework for study specific essential documents Organization and Access: Allows easy access to study documents with minimal effort Permits evaluation of the conduct of the study and assists to determine the quality of the data
Other Names for Regulatory Binder WHAT ARE SOME OTHER NAMES FOR THE REGULATORY BINDER? Derita Mom Dee Gram s
Regulatory Binder Names Regulatory Binder (RB) Reg Binder Investigator Binder Investigational Site File (ISF) Study Binder Master Trial File (MTF) Study Administrative Binder Regulatory Files Study File Trial Master File
Labeling the RB WHAT INFORMATION SHOULD BE INCLUDED IN THE LABELING OF THE RB?
Labeling the RB TITLE NUMBER THE BINDERS IF MORE THAN 1 (EX. 1 OF 3) PI NAME PROTOCOL NAME/IDENTIFIER IRB# SPONSOR
Responsibility WHOSE RESPONSIBILITY IS IT TO MAINTAIN THE RB?
Responsibility It may involve many different research study team members! Key Study Personnel (KSPs) Principal Investigator (PI) is ultimately responsible, but most often this task is delegated to other members of the study team, as well Investigators Clinical Research Coordinators Research Associates/Assistants Clinical Research Associates (Monitors) Data Managers Regulatory Affairs staff
Best Practices WHAT BEST PRACTICES RE: MAINTENANCE, ORGANIZATION, AND STORAGE DO YOU FOLLOW WHEN MAINTAINING THE RB?
RB Best Practices Maintenance: Establish the RB at the beginning of the study Keep the binder current, up-to-date, and organized Subject specific information/documents are usually maintained separately from the Regulatory Binder in subject specific research records Do not destroy earlier versions of essential documents Organization: Store items in reverse chronological order Customize the binder to the needs of the specific protocol Order the sections to facilitate easy referencing File sections that most referenced in the front of the binder Industry sponsored trials may have template binders to be used
Best Practices cont’d Storage: Store the binder in a safe and secure location at the investigator’s site Documents not stored in the RB should have a note (NTF)/memo indicating the location of where the documents will be maintained outside of the RB Maintain subject confidentiality It is suggested the following be maintained outside of the RB: Signed ICFs Test results Completed CRFs Screening/Enrollment log Master Log Contracts/budget/study financial records
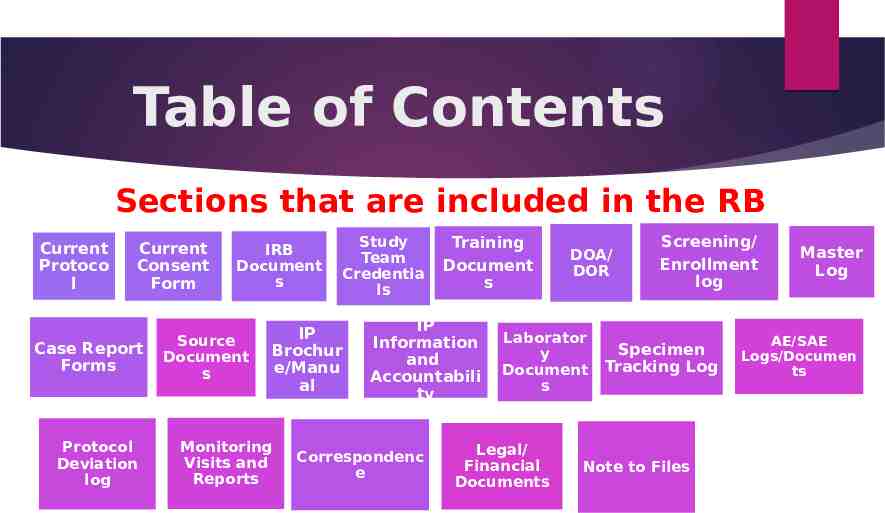
Table of Contents Sections that are included in the RB Current Protoco l Current Consent Form Case Report Forms Protocol Deviation log IRB Document s Source Document s Study Team Credentia ls IP Brochur e/Manu al Monitoring Visits and Reports Training Document s IP Information and Accountabili ty Correspondenc e DOA/ DOR Laborator y Document s Legal/ Financial Documents Screening/ Enrollment log Specimen Tracking Log Note to Files Master Log AE/SAE Logs/Documen ts
Current Protocol Most updated, current IRB approved protocol version PI signature page of the protocol. Why does the PI need to sign the signature page? Protocol Summary documents of changes if this is an amended version
Current Informed Consent Form (ICF) HOW DO YOU ENSURE THE STUDY TEAM HAS THE MOST UPDATED, IRB APPROVED CONSENT FORM AVAIALBLE FOR THEIR USE?
Signed ICFs WHERE DO YOU MAINTAIN SIGNED ICFs?
Institution al Review Board (IRB) PURPOSE: THIS SECTION IS MAINTAINED TO DOCUMENT THE TRIAL HAS OBTAINED IRB/IEC APPROVAL AND CAN BE CONDUCTED THE DOCUMENTS IN THIS SECTION CAN ALSO HELP TO IDENTIFY THE CURRENT VERSION OF IRB APPROVED DOCUMENTS
IRB WHAT DOCUMENTS SHOULD BE MAINTAINED IN THIS SECTION?
IRB cont’d IRB Submissions Initial/Ongoing/ Continuing/ Revision/ Amendment/ Termination Including all versions of the following: Protocol with signature page ICF Recruitment materials Blank CRFs Participant educational/study informational materials Reportable events (protocol deviations/ unanticipated problems/ SAEs/New information) Data Safety Monitoring Board Reports/Other Sponsor reports Correspondenc e with the IRB Including: Approval letters Proviso letters PI responses to IRB IRB notices of upcoming expiration dates Request for corrected IRB letter IRB stamped approved documents IRB Membershi p Roster Maintain from date of first submission to close out of the study (all 4 boards if UTHSC) IRB FWA
IRB cont’d WHAT TIPS CAN YOU GIVE ON HOW YOU MAINTAIN THIS SECTION OF YOUR RB?
Organizing the IRB Section Helpful Tips: Approval letter on top of the “clipped” submission Each submission may have a title page (colored paper) identifying the specifics of the submission/approval Type of submission, date submitted Date approved Summary of changes, if applicable Suggested to maintain IRB documents outside of iMedRIS
Study Team Credentials WHAT DOCUMENTS ARE MAINTAINED AS PART OF THE STUDY TEAM’S CREDENTIALS?
Study Team Credentials cont’d Curriculum vitae (CVs)/resume/bio sketch for the Principal Investigator/ sub-or co-investigator(s) and key study personnel (KSP) for the study Financial Disclosure Forms and Conflict of interest forms Clinical professional licensure/certification (dental, medical, nursing, pharmacy, etc.) for all KP Training documents (HSP, GCP, protocol, specific protocol procedures, etc.)
Study Team Credentials cont’d (1)WHERE DO YOU MAINTAIN THESE TYPES OF DOCUMENTS? (2)IF CREDENTIALS ARE MAINTAINED IN A CENTRAL/ELECTRONIC LOCATION, WHAT NEEDS TO OCCUR AT THE CLOSURE OF A STUDY?
Study Team Credentials Best Practices Maintain these credentials throughout the KSP’s participation in the study (and some studies require 2 years post study) The KSPs’ credentials should be maintained at the time (or before) their study participation to the end of their participation in the study (These dates should be in alliance with the DOAL) CV/Resumes should be current/signed/dated Industry standard is to update CV/Resume every 2 years WHY?
Training Documents NAME TYPES OF TRAINING REQUIRED FOR KSPs
Training Documents Human Subject Protection ICH GCP E6 (R2) FDA regulations 45CFR46 Declaration of Helsinki/Nuremburg Code/Belmont Report Institution specific training Protocol/procedures/IP In-services Study related meetings IATA/Biohazard SOPs/MOPs Site Initiation Visit cont’d
Delegation Log ADDITIONAL NAMES: DELEGATION OF AUTHORITY LOG (DOAL) DELEGATION OF AUTHORITY (DOA) DELEGATION OF RESPONSIBILITY LOG SIGNATURE LOG
DOA/DOAL/DOR NAME 4 PURPOSES OF THE DOAL PI PI KSPs
DOA/DOAL/DOR cont’d Purpose: Identifies the KSP involved in the study Matches KSP’s name to handwriting/initials Documents the delegated task(s) that have been assigned by the PI to the KSPs, ensure KSP is qualified to perform the delegated task Documents the timeline of an individual’s participation in the study
DOA/DOAL/DOR Best Practices: This document must remain current at all times Should be completed before/at the time the study is initiated The PI must initial and date any changes that are made throughout the life of the study He/she must also sign this form at the closure of the study cont’d
Purpose: Subject/ Participan t Logs Documentation of the individuals approached/screened and/or enrolled in the study Screening/ Enrollment Master log Do you have other names for these Essential documents?
Subject/ Participant Logs cont’d WHERE DO YOU USUALLY STORE THESE TYPES OF DOCUMENTS?
Screening/Enrollment Log Purpose: Captures patients that have been approached/screened to determine participation desire and initial eligibility To record the consent of all subjects wanting to participate and meets eligibility criteria Documents Provide potential subjects who where approached regarding their participation in the study a comprehensive list of all subjects who were screened for eligibility Documents chronological enrollment of subjects Documents the reason a potential subjects was not enrolled Maintains the status of the subject’s participation (withdrawal/termination/completed the study)
Subject Logs Best Practices Record subjects as they are screened/consented to ensure completeness and accuracy of the data, including screen failures This log should contain no identifying information Number each page At the conclusion of the study, identify the final page of the log
Master Log Purpose: Subject Identification Code List Confidential list of subject names/contact information
Case Report Forms (CRFs) Purpose: Documentation of protocol required information for each subject Facilitation of data collection and entry
Blank Case Report Forms (CRFs) CRFs include: Data collection sheets Questionnaires Worksheets A copy of blank CRFs should be maintained in the RB
Completed Case Report Forms WHERE DO YOU USUALLY STORE COMPLETED CRFs?
CRFs Best Practices All completed CRFs should be maintained as essential documents CRFs should be dated/signed by the study team member completing the form (ALCOA-C) Data reported on the CRF should be consistent with the source documents Any change or correction to a CRF should be dated, initialed, and explained (if necessary) and should not obscure the original entry
Source Documents WHERE ARE SOURCE DOCUMENTS MAINTAINED?
Source Document s cont’d Place of storage of source documents: Patient/Subject’s medical records Subject’s research record RB, for a small amount of participants
Investigational Product (IP) Purpose: Documentation of the receipt and maintenance of the IP Documents to be maintained: Investigational brochure, package insert, or device manual, all versions Sample of product label Documentation of study product shipment/receipt/dispensing/return/disposal Include dates, quantities, batch/serial #, expiration dates Instructions for IP handling Randomization instructions, if applicable
IP cont’d Documents to be maintained: Temperature log, when applicable Storage/maintenance instructions Unblinding procedures, as applicable
Laboratory/Testing Center Documents Purpose: Documentation of the normal ranges for the laboratory tests/procedures/laboratory’s credentials as being a qualified lab to test specimens collected for the study Documents to be maintained: Copy of laboratory certification and accreditations Laboratory normal reference ranges/values for procedures included in the protocol Certification of analysis/reliability of tests performed, when requested Copy of Laboratory/Testing Center’s Director’s license and CV (current within 2 years)
Specimen Tracking Log Purpose: Provides a means to tracks specimens/samples collected as study procedures Types of samples to track: Specimen Tracking Log Retained tissue log Documents to maintain Shipping/Receipt/Maintenance documentation Storage temperature logs
AEs/ SAEs Purpose: Documentation of side effects/adverse events (AEs)/serious adverse events (SAEs)/unexpected adverse events experienced by a subject
AEs/SAEs/ Unanticipated Events Maintain the following: Log for each subject, if applicable Log for study Forms/CAPAs Correspondence, copies and acknowledgement of reports of internal AEs External reports to IRB, Sponsor, regulatory authorities IND Safety Reports
Protocol Deviations Purpose: Documentation of non-compliance with the protocol Best Practices: Maintain a protocol deviation log Documentation/memo/Corrective Action Preventive Action (CAPA), as applicable for each deviation Record events a soon as possible after the event, ALCOA-C
Monitoring Visits Purpose: Documentation of the monitoring visit/visit type/ results of visit(s)/findings(s) Maintain the following in this section: Monitoring reports/letters/Correspondence Monitoring Log: Each Visit should be documented by the monitor and a study team member by dating and signing the log The reason for each visit should also be document on the log Pre-study visit, site initiation, routine visits, close-out visit, etc.
Correspondence Purpose To record discussions/ correspondence that are significant to the conduct of the study resulting in a complete and accurate audit trail Types of Correspondence (e-mails/faxes/letters/electronic documents, meeting notes, phone calls) to maintain from: Sponsor Contract Research Organization (CROs) Monitor IRB (prefer to keep this correspondence with the IRB section) Study Team Correspondence regarding individual subjects should/may be maintained with their research records
Legal/Financial Documents WHERE DO YOU MAINTAIN LEGAL/FINANCIAL DOCUMENTS?
Legal/Financial Documents cont’d FDA documents Contracts Budgets Subject payments Letter of Understanding/Confidentiality Agreement Data Sharing Agreement Material Transfer Agreement Signed agreements between parties (i.e., sponsor/CRO/Institution investigators)
Note to Files (NTFs) WHERE CAN NTFs BE MAINTAINED?
NTFs Purpose: To document the reason for missing/delayed/incorrect information/any discrepancies To document the corrective action taken/resolution of discrepancies To document the location of documents stored outside of the RB To provide additional information or clarification cont’d NTFs should include the following: Signature of the author and the date created and the PI (when applicable) Description of the event (who, what, where, when, how, why) Actions taken to resolve the issue Any necessary CAPA Pertinent information to provide clarification of an event
Conclusion The RB should: Be maintained in an up-to-date manner at all times Organized in a logical manner Located in a central location and accessible to the appropriate study team members Be maintained in paper / electronic/or combination of both formats
Any Questions or Comments?





