Mass Spectrometry-Based Methods for Protein Identification Joseph A.
























66 Slides6.93 MB
Mass Spectrometry-Based Methods for Protein Identification Joseph A. Loo Department of Biological Chemistry David Geffen School of Medicine Department of Chemistry and Biochemistry University of California Los Angeles, CA USA
Genomics and Proteomics Characterizing many genes and gene products simultaneously
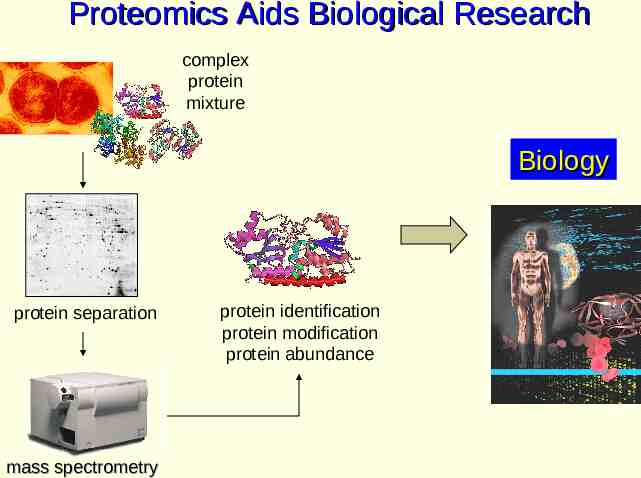
Proteomics Aids Biological Research complex protein mixture Biology protein separation mass spectrometry protein identification protein modification protein abundance
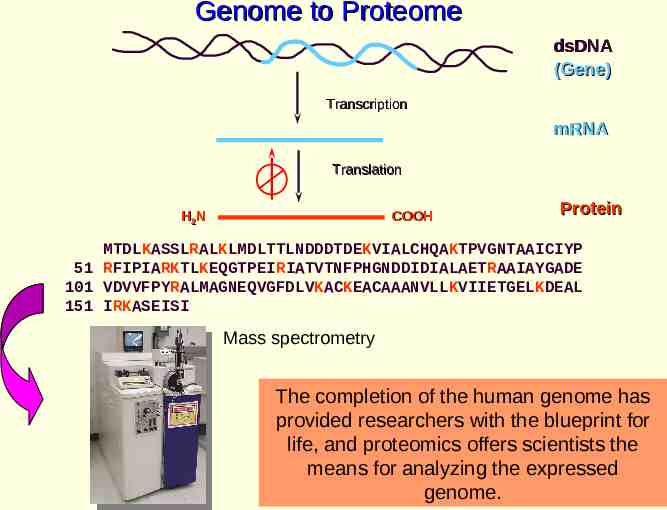
Proteomics - What is it? An assay to systematically analyze the diverse properties of proteins Biological processes are dynamic A quantitative comparison of states is required The study of protein expression and function on a genome scale Purpose: Examine altered gene expression pathways in disease states and under different environmental conditions The completion of the human genome has provided researchers with the blueprint for life, and proteomics offers scientists the means for analyzing the expressed genome.
Genome to Proteome dsDNA (Gene) Transcription mRNA Translation COOH H2N Protein MTDLKASSLRALKLMDLTTLNDDDTDEKVIALCHQAKTPVGNTAAICIYP 51 RFIPIARKTLKEQGTPEIRIATVTNFPHGNDDIDIALAETRAAIAYGADE 101 VDVVFPYRALMAGNEQVGFDLVKACKEACAAANVLLKVIIETGELKDEAL 151 IRKASEISI Mass spectrometry The completion of the human genome has provided researchers with the blueprint for life, and proteomics offers scientists the means for analyzing the expressed genome.
Approaches for Protein Identification What is this protein? Molecular weight Isoelectric point Amino acid composition Other physical/chemical characteristics Partial or complete amino acid sequence Edman (N-terminal sequence) - if N-term. not blocked C-terminal sequence - not commonly performed Mass spectrometry-measured information
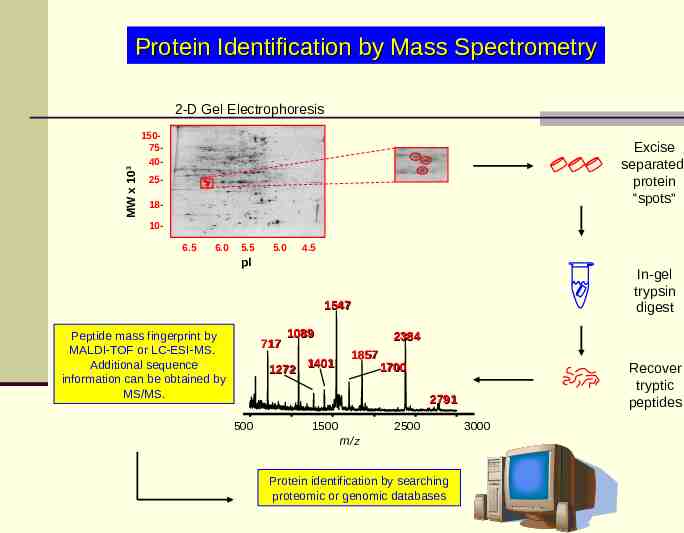
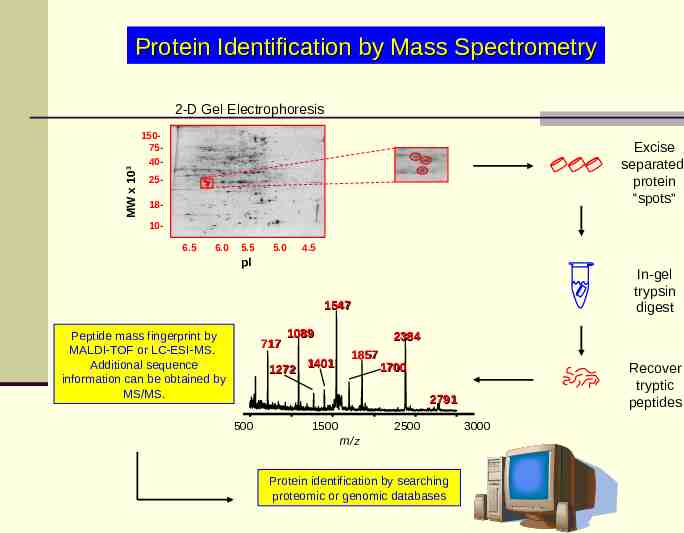
Protein Identification by Mass Spectrometry MW x 103 2-D Gel Electrophoresis 1507540- Excise separated protein “spots” 2518106.5 6.0 5.5 5.0 4.5 pI In-gel trypsin digest 1547 Peptide mass fingerprint by MALDI-TOF or LC-ESI-MS. Additional sequence information can be obtained by MS/MS. 717 1089 1401 1272 2384 1857 1700 Recover tryptic peptides 2791 500 2500 1500 m/z Protein identification by searching proteomic or genomic databases 3000
Mass Spectrometry: A method to “weigh” molecules Other information can be inferred from a weight measurement. Post-translational modifications Molecular interactions Shape Sequence Physical dimensions etc. A simple measurement of mass is used to confirm the identity of a molecule, but it can be used for much more
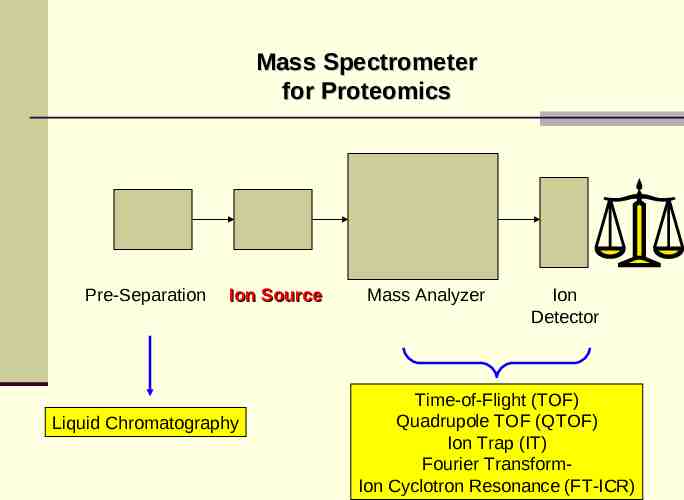
Mass Spectrometer for Proteomics Pre-Separation Ion Source Liquid Chromatography Mass Analyzer Ion Detector Time-of-Flight (TOF) Quadrupole TOF (QTOF) Ion Trap (IT) Fourier TransformIon Cyclotron Resonance (FT-ICR)
The Nobel Prize in Chemistry 2002 "for the development of methods for identification and structure analyses of biological macromolecules" "for their development of soft desorption ionisation methods for mass spectrometric analyses of biological macromolecules" John Fenn Koichi Tanaka
Electrospray: Generation of aerosols and droplets
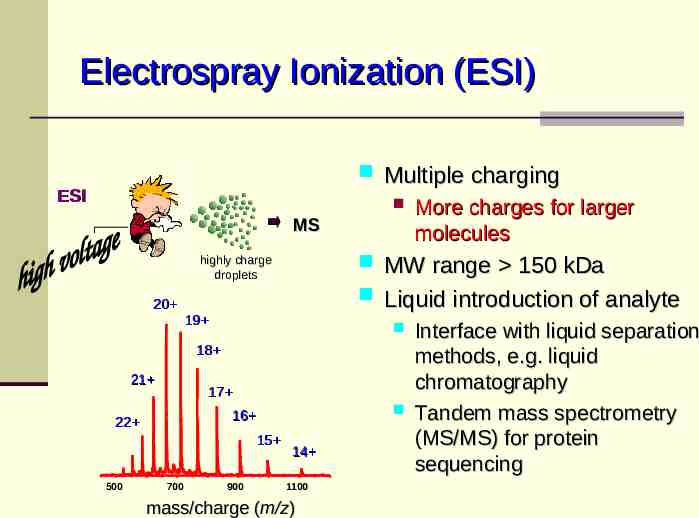
Electrospray Ionization (ESI) ESI MS highly charge droplets 20 19 18 21 17 16 22 15 500 700 900 14 1100 mass/charge (m/z) Multiple charging More charges for larger molecules MW range 150 kDa Liquid introduction of analyte Interface with liquid separation methods, e.g. liquid chromatography Tandem mass spectrometry (MS/MS) for protein sequencing
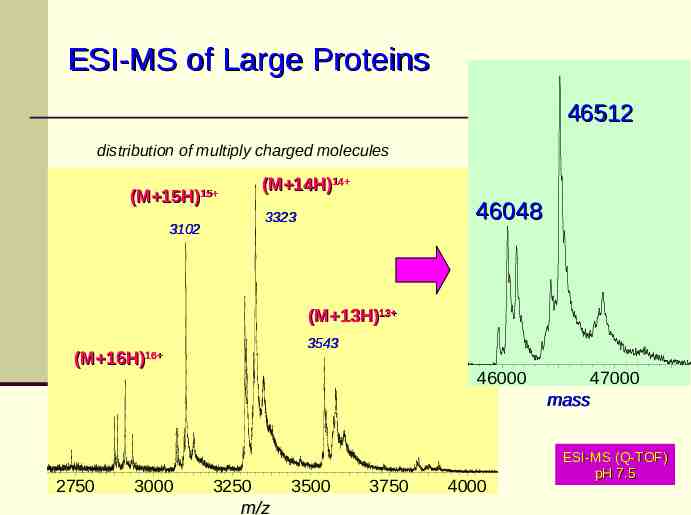
IGF 1R, fraction 1 Operator: JL Conditions: NH4bicarb Instrument: Q-TOF Collision Energy: 4 ESI-MS of Large100Proteins 46512 distribution of multiply charged molecules 1R, fraction 1 rator: JL ditions: NH4bicarb Instrument: Q-TOF Collision Energy: 4 100 (M 15H) 15 3102 (M 14H)14 3323 46048 % (M 13H)13 % 3543 (M 16H) 16 0 9-JUL-2001 Laboratory: PGRD - Discovery Technologies Cone (V): 150 2750 3000 0 3250 m/z 3500 45000 3750 46000 4000 47000 mass ESI-MS (Q-TOF) pH 7.5 m/z 48
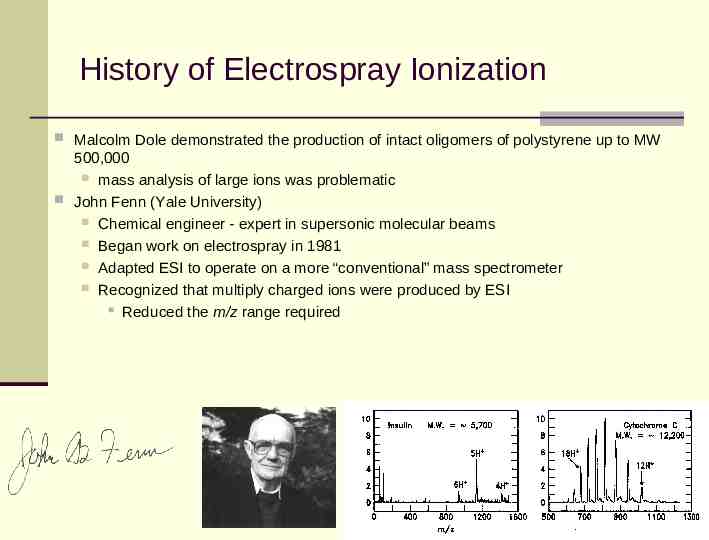
History of Electrospray Ionization Malcolm Dole demonstrated the production of intact oligomers of polystyrene up to MW 500,000 mass analysis of large ions was problematic John Fenn (Yale University) Chemical engineer - expert in supersonic molecular beams Began work on electrospray in 1981 Adapted ESI to operate on a more “conventional” mass spectrometer Recognized that multiply charged ions were produced by ESI Reduced the m/z range required
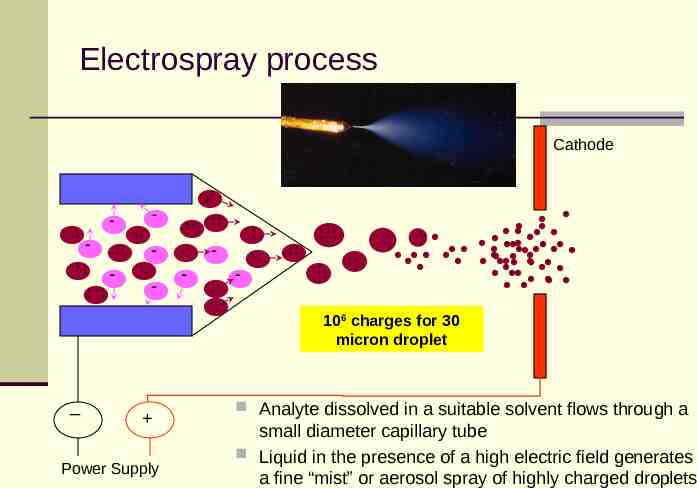
Electrospray process Cathode - - - - - - Power Supply - - 106 charges for 30 micron droplet Analyte dissolved in a suitable solvent flows through a small diameter capillary tube Liquid in the presence of a high electric field generates a fine “mist” or aerosol spray of highly charged droplets
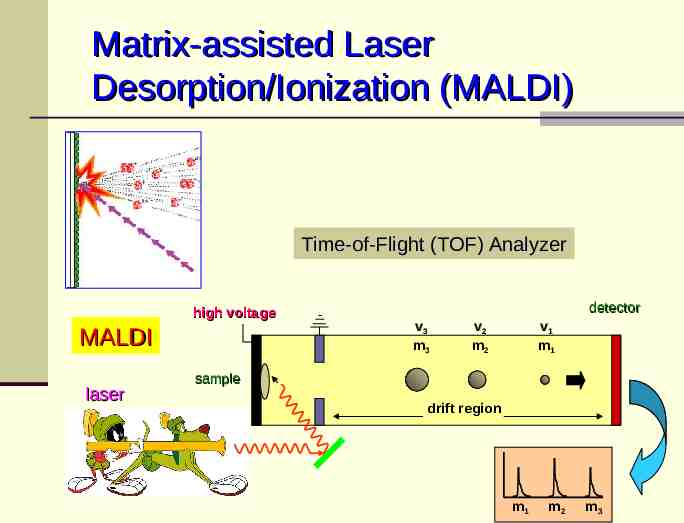
Matrix-assisted Laser Desorption/Ionization (MALDI) Time-of-Flight (TOF) Analyzer high voltage MALDI laser detector v3 m3 v2 m2 v1 m1 sample drift region m1 m2 m3
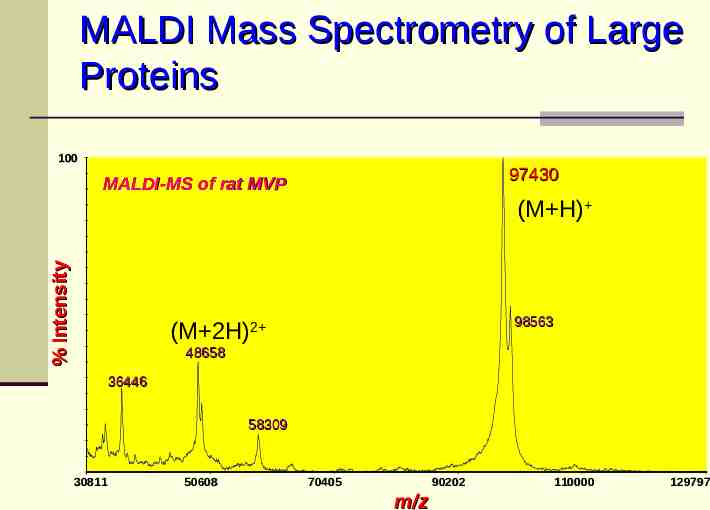
MALDI Mass Spectrometry of Large Proteins 100 97430 MALDI-MS of rat MVP % Intensity (M H) 98563 (M 2H)2 48658 36446 58309 30811 50608 70405 90202 m/z 110000 129797
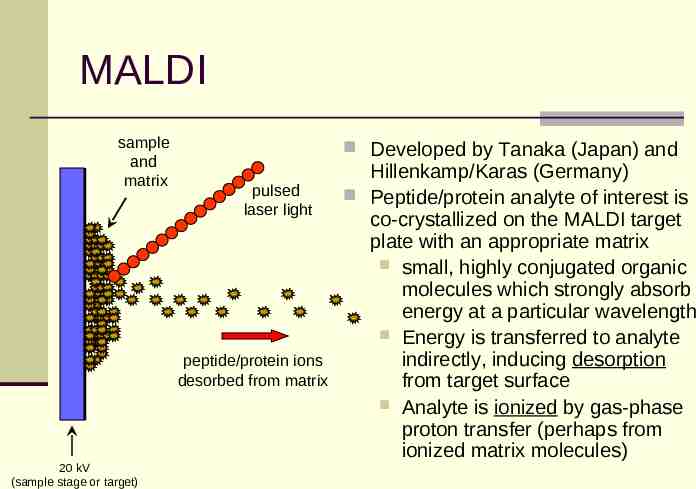
MALDI sample and matrix Developed by Tanaka (Japan) and pulsed laser light peptide/protein ions desorbed from matrix 20 kV (sample stage or target) Hillenkamp/Karas (Germany) Peptide/protein analyte of interest is co-crystallized on the MALDI target plate with an appropriate matrix small, highly conjugated organic molecules which strongly absorb energy at a particular wavelength Energy is transferred to analyte indirectly, inducing desorption from target surface Analyte is ionized by gas-phase proton transfer (perhaps from ionized matrix molecules)
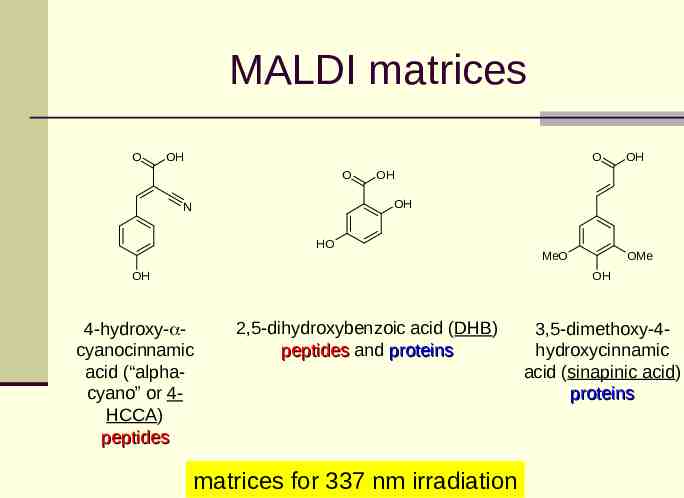
MALDI matrices O OH O O OH OH OH N HO OH OMe MeO OH 4-hydroxy- cyanocinnamic acid (“alphacyano” or 4HCCA) peptides 2,5-dihydroxybenzoic acid (DHB) peptides and proteins matrices for 337 nm irradiation 3,5-dimethoxy-4hydroxycinnamic acid (sinapinic acid) proteins
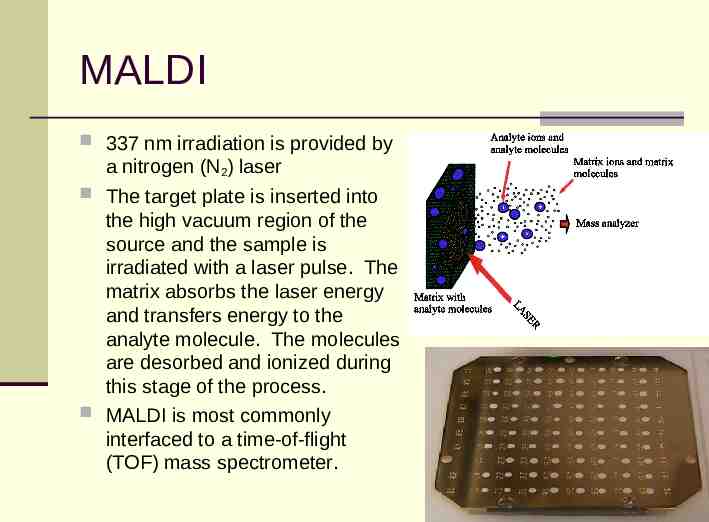
MALDI 337 nm irradiation is provided by a nitrogen (N2) laser The target plate is inserted into the high vacuum region of the source and the sample is irradiated with a laser pulse. The matrix absorbs the laser energy and transfers energy to the analyte molecule. The molecules are desorbed and ionized during this stage of the process. MALDI is most commonly interfaced to a time-of-flight (TOF) mass spectrometer.
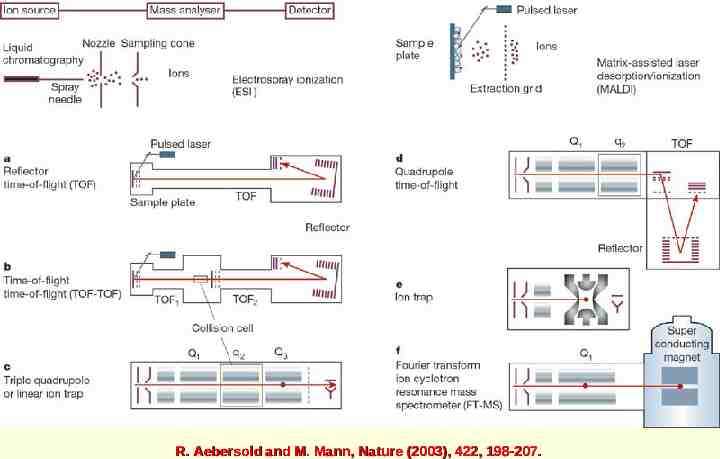
R. Aebersold and M. Mann, Nature (2003), 422, 198-207.
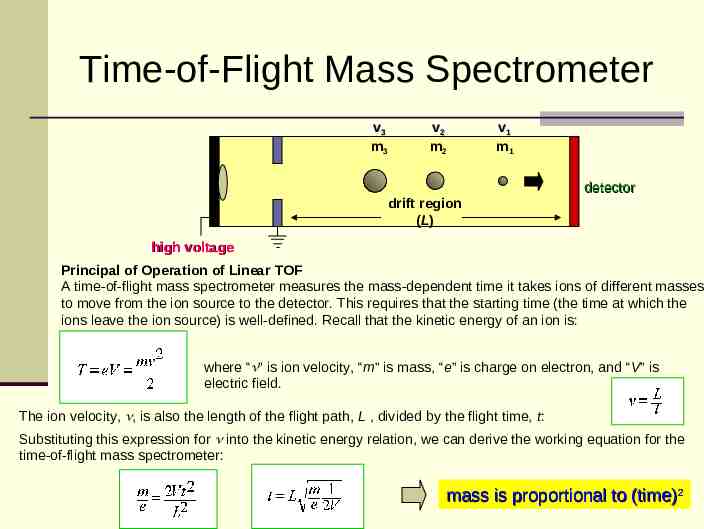
Time-of-Flight Mass Spectrometer v3 m3 v2 m2 v1 m1 detector drift region (L) high voltage Principal of Operation of Linear TOF A time-of-flight mass spectrometer measures the mass-dependent time it takes ions of different masses to move from the ion source to the detector. This requires that the starting time (the time at which the ions leave the ion source) is well-defined. Recall that the kinetic energy of an ion is: where “ ” is ion velocity, “m” is mass, “e” is charge on electron, and “V” is electric field. The ion velocity, , is also the length of the flight path, L , divided by the flight time, t: Substituting this expression for into the kinetic energy relation, we can derive the working equation for the time-of-flight mass spectrometer: mass is proportional to (time)2
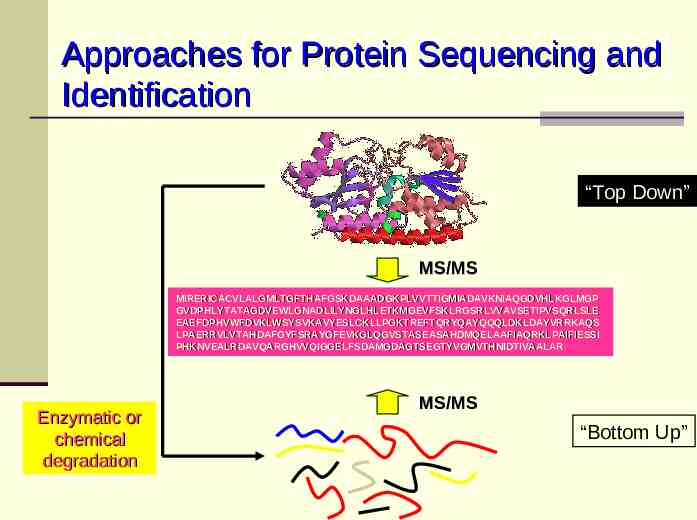
Approaches for Protein Sequencing and Identification “Top Down” MS/MS MIRERICACVLALGMLTGFTHAFGSKDAAADGKPLVVTTIGMIADAVKNIAQGDVHLKGLMGP GVDPHLYTATAGDVEWLGNADLILYNGLHLETKMGEVFSKLRGSRLVVAVSETIPVSQRLSLE EAEFDPHVWFDVKLWSYSVKAVYESLCKLLPGKTREFTQRYQAYQQQLDKLDAYVRRKAQS LPAERRVLVTAHDAFGYFSRAYGFEVKGLQGVSTASEASAHDMQELAAFIAQRKLPAIFIESSI PHKNVEALRDAVQARGHVVQIGGELFSDAMGDAGTSEGTYVGMVTHNIDTIVAALAR Enzymatic or chemical degradation MS/MS “Bottom Up”
Identification of proteins from gels Proteins are separated first by high resolution two-dimensional polyacrylamide gel electrophoresis and then stained. At this point, to identify an individual or set of protein spots, several options can be considered by the researcher, depending on availability of techniques. For protein spots that appear to be relatively abundant (e.g., more than 1 pmol), traditional protein characterization methods may be employed. employed Methods such as amino acid analysis and Edman sequencing can be used to provide necessary protein identification information. With 2-DE, approximate molecular weight and isoelectric point characteristics are provided. Augmented with information on amino acid composition and/or amino-terminal sequence, a confident identification can be obtained. The sensitivity gains of using MS allows for the identification of proteins below the one pmol level and in many cases in the femtomole regime.
Protein Identification by Mass Spectrometry MW x 103 2-D Gel Electrophoresis 1507540- Excise separated protein “spots” 2518106.5 6.0 5.5 5.0 4.5 pI In-gel trypsin digest 1547 Peptide mass fingerprint by MALDI-TOF or LC-ESI-MS. Additional sequence information can be obtained by MS/MS. 717 1089 1401 1272 2384 1857 1700 Recover tryptic peptides 2791 500 2500 1500 m/z Protein identification by searching proteomic or genomic databases 3000
Protein Cleavage For the application of mass spectrometry for protein identification, the protein bands/spots from a 2-D gel are excised and are exposed to a highly specific enzymatic cleavage reagent (e.g., trypsin cleaves on the C-terminal side of arginine and lysine residues). The resulting tryptic fragments are extracted from the gel slice and are then subjected to MS-methods. One of the major barriers to high throughput in the proteomic approach to protein identification is the “in-gel” proteolytic digestion and subsequent extraction of the proteolytic peptides from the gel. Common protocols for this process are often long and labor intensive. protein digestion robot
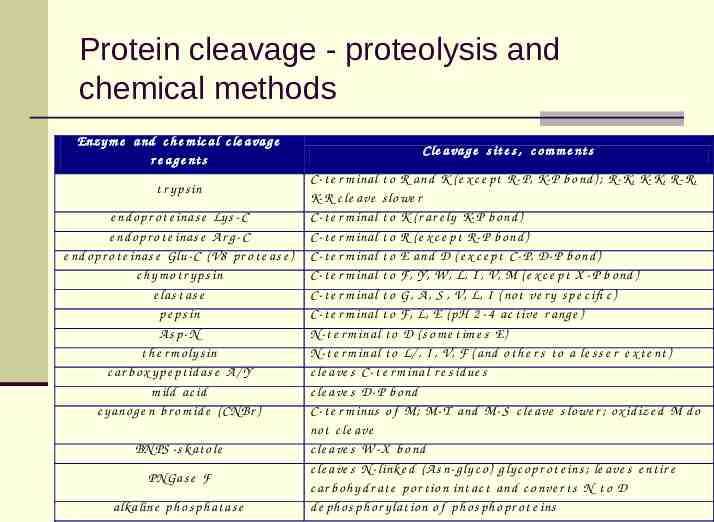
Protein cleavage - proteolysis and chemical methods Enz y m e a nd c h e m ic a l c le a va g e r e a g e nt s t r y p s in e nd o p r o t e inas e Ly s - C e nd o p r o t e inas e A r g - C e nd o p r o t e inas e Glu- C ( V8 p r o t e as e ) c h y m o t r y p s in e las t as e p e ps in As p-N t h e r m o ly s in c ar b o x y p e p t id as e A / Y m ild ac id c y ano g e n b r o m id e ( CN Br ) BN PS - s k at o le PNGas e F alk aline p h o s ph at as e C le a va g e s it e s , c o m m e nt s C- t e r m inal t o R and K ( e x c e p t R- P, K- P b o nd ) ; R- K, K- K, R- R, K- R c le ave s lo we r C- t e r m inal t o K ( r ar e ly K- P b o nd ) C- t e r m inal t o R ( e x c e p t R- P b o nd ) C- t e r m inal t o E and D ( e x c e p t C - P, D- P b o nd ) C- t e r m inal t o F, Y, W , L, I , V, M ( e x c e p t X - P b o nd ) C- t e r m inal t o G, A , S , V, L, I ( no t ve r y s p e c ifi c ) C- t e r m inal t o F, L, E ( pH 2 - 4 ac t ive r ang e ) N - t e r m inal t o D ( s o m e t im e s E) N - t e r m inal t o L/ , I , V, F ( and o t h e r s t o a le s s e r e x t e nt ) c le a ve s C- t e r m inal r e s id ue s c le a ve s D- P b o nd C- t e r m inus o f M; M- T and M- S c le ave s lo we r ; o x id ize d M d o no t c le ave c le a ve s W - X b o nd c le a ve s N - link e d ( A s n- g ly c o ) g ly c o p r o t e ins ; le ave s e nt ir e c ar b o h y d r at e po r t io n int ac t and c o nve r t s N t o D d e ph o s ph o r y lat io n o f ph o s p h o p r o t e ins
Mass spectrometry-based protein identification A mass spectrum of the resulting digest products produces a “peptide map” or a “peptide fingerprint”. The measured masses can be compared to theoretical peptide maps derived from database sequences for identification. There are a few choices of mass analysis that can be selected from this point, depending on available instrumentation and other factors. The resulting peptide fragments can be subjected to MALDI-MS or ESIMS analysis. A small aliquot of the digest solution can be directly analyzed by MALDIMS to obtain a peptide map. The resulting sequence coverage (relative to the entire protein sequence) displayed from the total number of tryptic peptides observed in the MALDI mass spectrum can be quite high, i.e., greater than 80% of the sequence, although it can vary considerably depending on the protein, sample amount, etc. The measured molecular weights of the peptide fragments along with the specificity of the enzyme employed can be searched and compared against protein sequence databases using a number of computer searching routines available on the Internet.
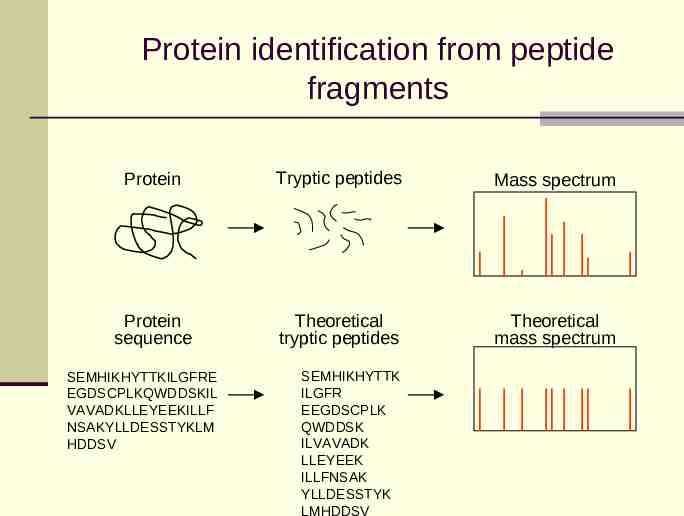
Protein identification from peptide fragments Protein Tryptic peptides Mass spectrum Protein sequence Theoretical tryptic peptides Theoretical mass spectrum SEMHIKHYTTKILGFRE EGDSCPLKQWDDSKIL VAVADKLLEYEEKILLF NSAKYLLDESSTYKLM HDDSV SEMHIKHYTTK ILGFR EEGDSCPLK QWDDSK ILVAVADK LLEYEEK ILLFNSAK YLLDESSTYK LMHDDSV
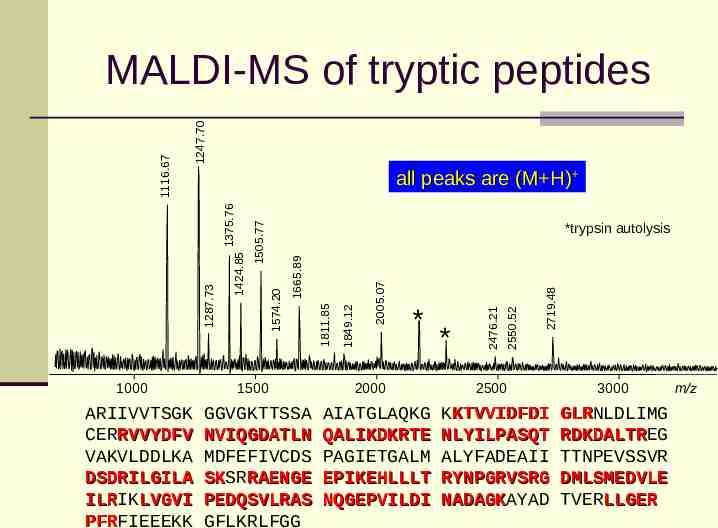
1247.70 ARIIVVTSGK CERRVVYDFV VAKVLDDLKA DSDRILGILA ILRIK ILR LVGVI PFRFIEEEKK PFR 1500 GGVGKTTSSA NVIQGDATLN MDFEFIVCDS SKSR SK RAENGE PEDQSVLRAS GFLKRLFGG 2000 AIATGLAQKG QALIKDKRTE PAGIETGALM EPIKEHLLLT NQGEPVILDI 2719.48 * 2550.52 * 2476.21 2005.07 1849.12 1811.85 1665.89 *trypsin autolysis 1574.20 1424.85 1287.73 1000 1505.77 all peaks are (M H) 1375.76 1116.67 MALDI-MS of tryptic peptides 2500 KKTVVIDFDI NLYILPASQT ALYFADEAII RYNPGRVSRG NADAGKAYAD NADAGK 3000 GLRNLDLIMG GLR RDKDALTREG RDKDALTR TTNPEVSSVR DMLSMEDVLE TVERLLGER m/z
ESI-MS and LC-MS for protein identification An approach for peptide mapping similar to MALDI-MS uses ESI-MS. A peptide map can be obtained by analysis of the peptide mixture by ESI-MS. An advantage of ESI is its ease of coupling to separation methodologies such as HPLC. Thus, alternatively, to reduce the complexity of the mixture, the peptides can be separated by HPLC with subsequent mass measurement by on-line ESIMS. The measured masses can be compared to sequence databases. 100 Rel. Abund. 9.4 8.4 0 Rel. Abund. 100 6.2 5 6 6.8 7 7.7 8.9 8 9 9.8 LC-MS with ESI 10 11 12 Time (min) 965.3 (M 2H)2 MW 1928.6 Da 629.0 0 400 800 1200 1600 2000 m/z
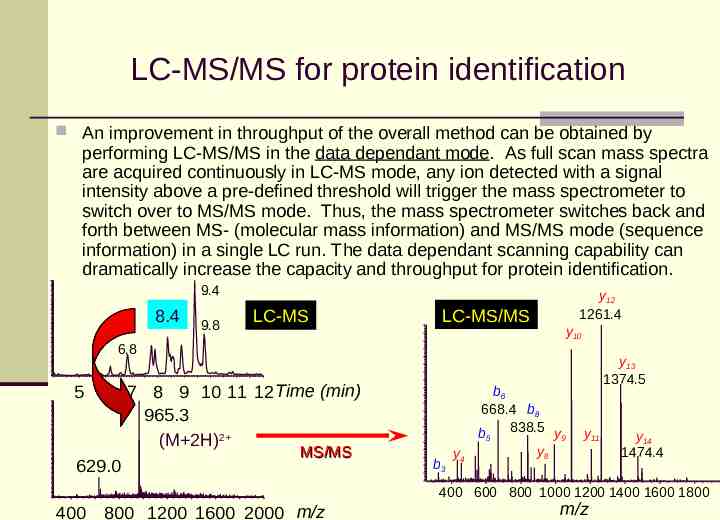
LC-MS/MS for protein identification An improvement in throughput of the overall method can be obtained by performing LC-MS/MS in the data dependant mode. As full scan mass spectra are acquired continuously in LC-MS mode, any ion detected with a signal intensity above a pre-defined threshold will trigger the mass spectrometer to switch over to MS/MS mode. Thus, the mass spectrometer switches back and forth between MS- (molecular mass information) and MS/MS mode (sequence information) in a single LC run. The data dependant scanning capability can dramatically increase the capacity and throughput for protein identification. 9.4 8.4 9.8 LC-MS LC-MS/MS y12 1261.4 y10 6.8 5 6 7 8 9 10 11 12Time (min) 965.3 (M 2H)2 MS/MS 629.0 b3 y4 b6 668.4 b8 b5 838.5 y9 y8 y13 1374.5 y11 y14 1474.4 400 600 800 1000 1200 1400 1600 1800 400 800 1200 1600 2000 m/z m/z
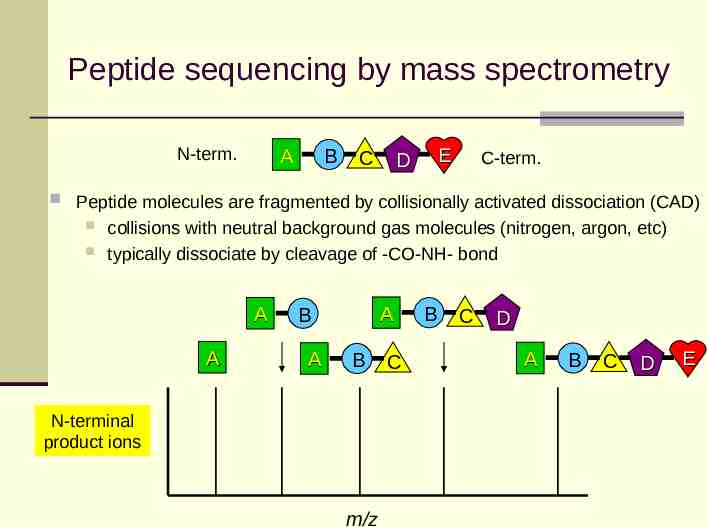
Peptide sequencing by mass spectrometry N-term. A B C E D C-term. Peptide molecules are fragmented by collisionally activated dissociation (CAD) collisions with neutral background gas molecules (nitrogen, argon, etc) typically dissociate by cleavage of -CO-NH- bond A A A B A B N-terminal product ions m/z C B C D A B C D E
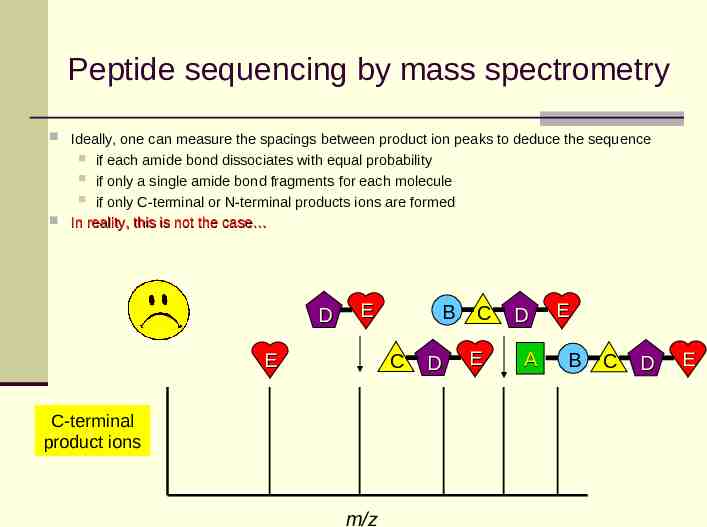
Peptide sequencing by mass spectrometry Ideally, one can measure the spacings between product ion peaks to deduce the sequence if each amide bond dissociates with equal probability if only a single amide bond fragments for each molecule if only C-terminal or N-terminal products ions are formed In reality, this is not the case D E E B C C-terminal product ions m/z D C E D A E B C D E
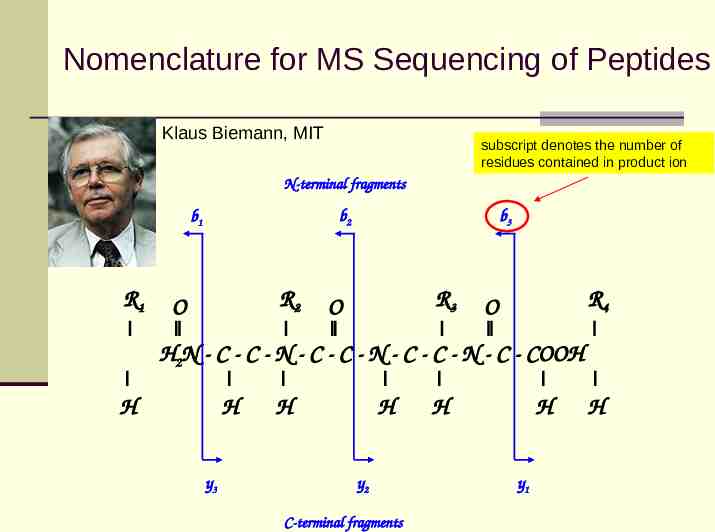
Nomenclature for MS Sequencing of Peptides Klaus Biemann, MIT subscript denotes the number of residues contained in product ion N-terminal fragments b2 b1 R1 R2 O b3 R3 O R4 O H2N - C - C - N - C - C - N - C - C - N - C - COOH H H y3 H H y2 C-terminal fragments H H y1 H
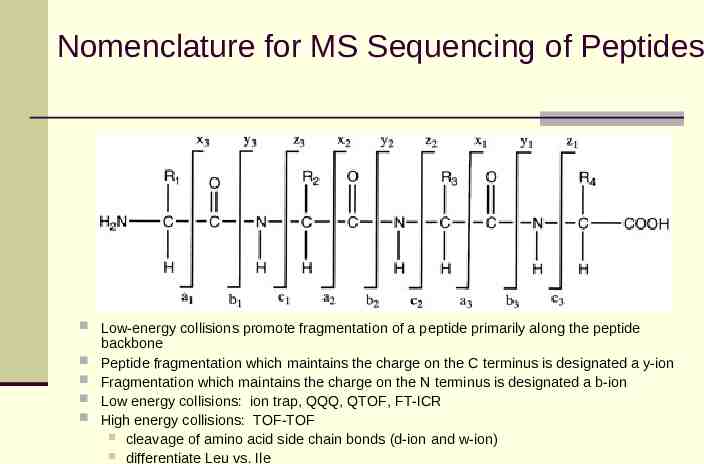
Nomenclature for MS Sequencing of Peptides Low-energy collisions promote fragmentation of a peptide primarily along the peptide backbone Peptide fragmentation which maintains the charge on the C terminus is designated a y-ion Fragmentation which maintains the charge on the N terminus is designated a b-ion Low energy collisions: ion trap, QQQ, QTOF, FT-ICR High energy collisions: TOF-TOF cleavage of amino acid side chain bonds (d-ion and w-ion) differentiate Leu vs. Ile
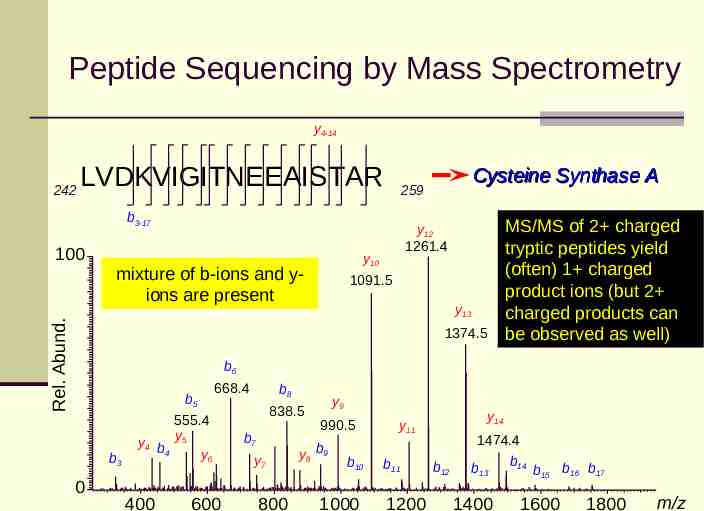
Peptide Sequencing by Mass Spectrometry y4-14 242 LVDKVIGITNEEAISTAR b3-17 y12 1261.4 y10 mixture of b-ions and yions are present Rel. Abund. 100 Cysteine Synthase A 259 1091.5 y13 1374.5 MS/MS of 2 charged tryptic peptides yield (often) 1 charged product ions (but 2 charged products can be observed as well) b6 b5 b3 0 668.4 555.4 y y4 b 5 4 y6 400 600 b8 838.5 b7 y7 800 y9 990.5 y 8 b9 b10 1000 y14 y11 b11 1200 b12 1474.4 b14 b13 b15 b16 b17 1400 1600 1800 m/z
Computer-based Sequence Searching Strategies A list of experimentally determined masses is compared to lists of computer-generated theoretical masses prepared from a database of protein primary sequences. With the current exponential growth in the generation of genomic data, these databases are expanding every day. There are typically three types of search strategies employed: searching with peptide fingerprint data searching with sequence data searching with raw MS/MS data. One limiting factor that must be considered for all of the approaches is that they can only identify proteins that have been identified and reside within an available database, or very homologous to one that resides in the database.
Searching with Peptide Fingerprints The majority of the available search engines allow one to define certain experimental parameters to optimize a particular search. Minimum number of peptides to be matched Allowable mass error Monoisotopic versus average mass data Mass range of starting protein Type of protease used for digestion Information about potential protein modification, such as N- and C-terminal modification, carboxymethylation, oxidized methionines, etc.
Searching with Peptide Fingerprints Most protein databases contain primary sequence information only Any shift in mass incorporated into the primary sequence as a result of post-translational modification will result in an experimental mass that is in disagreement with the theoretical mass. Modifications such as glycation and phosphorylation can result in missed identifications. A single amino acid substitution can shift the mass of a peptide to such a degree that even a protein with a great deal of homology with another in the database can not be identified.
Searching with Peptide Fingerprints A number of factors affect the utility of peptide fingerprinting. The greater the experimental mass accuracy, accuracy the narrower you can set your search tolerances, thereby increasing your confidence in the match, and decreasing the number of "false positive" responses. A common practice used to increase mass accuracy in peptide fingerprinting is to employ an autolysis fragment from the proteolytic enzyme as an internal standard to calibrate a MALDI mass spectrum. Peptide fingerprinting is also amenable to the identification of proteins in complex mixtures. Peptides generated from the digest of a protein mixture will simply return two or more results that are a "good" fit. Peptides that are "left over" in a peptide fingerprint after the identification of one component can be resubmitted for the possible identification of another component.
Web addresses of some representative internet resources for protein identification from mass spectrometry data Pr o g r a m W e b Ad d r e s s BLA S T h t t p:/ / www.e b i.ac .uk / b las t all/ Mas c o t h t t p :/ / www.m at r ix s c ie nc e .c o m / c g i/ ind e x .p l? p ag e / h o m e .h t m l Mas s S e ar c h h t t p :/ / c b r g .inf .e t h z .c h / S e r ve r / S e r ve r Bo o k le t / Mas s S e ar c h Ex .h t m l MO W S E h t t p :/ / s r s .h g m p .m r c .ac .uk / c g i- b in/ m o ws e h t t p :/ / www.nar r ad o r .e m b l- Pe pt id e S e ar c h h e id e lb e r g .d e / Gr o up Pag e s / Pag e Link / p e p t id e s e ar c h pag e .h t m l Pr o t e in Pr o s p e c t o r h t t p :/ / p r o s p e c t o r .uc s f .e d u/ Pr o wl h t t p :/ / p r o wl.r o c ke f e lle r .e d u/ S EQ UES T h t t p:/ / fi e ld s .s c r ip ps .e d u/ s e q ue s t /
Mascot Among the first programs for identifying proteins by peptide mass fingerprinting, MOWSE, developed out of a collaboration between Imperial Cancer Research Fund (ICRF) and SERC Daresbury Laboratory, UK. The name chosen was an acronym of Molecular Weight Search. The MOWSE databases were fully indexed so as to allow very rapid searching and retrieval of sequence data. Subsequently, the software was further developed and renamed Mascot. Licensed and distributed by Matrix Science Ltd. Specialized tools include Peptide Mass Fingerprint, Sequence Query, and MS/MS Ion Search. Search output Web-based. Good visual representation of search quality (graphical probability chart). Simple graphical user interface. Reports MOWSE scores as a quantitative measure of search quality.
Mowse Scoring Rather than just counting the number of matching peptides, Mowse uses empirically determined factors to assign a statistical weight to each individual peptide match. match Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 3:327, 1993.) Scoring scheme assigns more weight to matches of higher molecular weight peptides (more discriminating). Compensates for the non-random distribution of fragment molecular weights in proteins of different sizes. Was first protein identification program to recognize that the relative abundance of peptides of a given length in a proteolytic digest depends on the lengths of both peptide and protein. Developed for MALDI peptide mass fingerprinting. Probability-Based Mowse Mascot incorporates a probability-based enhanced Mowse algorithm, described in Perkins et al. (Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20:3551-3567, 1999). A simple rule can be used to judge whether a result is significant or not. Different types of matching (peptide masses and fragment ions) can be combined in a single search.
Databases Three components are required for database searching support of proteomics: MALDI or MS/MS data, the algorithms used to search protein databases with the MALDI or MS/MS data, and the protein databases themselves. The protein databases can be as small as one protein, can be large, public domain databases of all known and predicted proteins, or may be predicted open reading frames based on genomic sequence. A major challenge for database searching is that these protein databases are constantly changing, making database search results potentially obsolete as new entries are added that better fit the MALDI or MS data. Even as genomes are completed there is still flux as new coding regions are identified and novel mechanisms of increased translational complexity are better understood, such as alternative splice products, RNA editing, and ribosome slippage leading to novel, unexpected translation products.
Databases NCBI non-redundant (NCBInr) Non-redundant database from the National Center for Biotechnology Information for use with their search tools BLAST and Entrez; comprised of translated sequences from the Genbank /EMBL/DDBJ consortium, SwissProt, Protein Information Resource (PIR), and Brookhaven Protein Data Bank (PDB). New releases are published bimonthly while updates occur daily. OWL OWL is comprised of Swiss-Prot, PIR, translated Genbank, and NRL-3D (PDB). All sequences are compared to Swiss-Prot to remove identical and “trivially different “ sequences. Has not been updated since May, 1999. SWISSPROT While SwissProt contains only a subset of proteins, the proteins in this database are much better annotated and the sequences are much more reliable than those available in any other database. MSDB Comprehensive, non-identical protein sequence database maintained by the Proteomics Department at the Hammersmith Campus of Imperial College London. Designed specifically for MS applications.
Databases EST Clusters (dBEST) Division of GenBank that contains "single-pass" cDNA sequences, or Expressed Sequence Tags (EST’s), from a number of organisms. EST’s are relatively short, usually 3’ end sequences from isolated mRNA. EST’s tend to be highly redundant and the sequence is much lower quality than from other sources. An advantage to using these EST’s is that they represent only expressed sequences (no introns) and include alternative splice variants; their length, redundancy, and low quality are far improved by using clustered EST’s, such as the Compugen clusters. The EST database has some redundancy because it contains all possible combinations of alternative splice products, and so it can be very large (and slow to search). During a Mascot search, the nucleic acid sequences are translated in all six reading frames. dbEST is a very large database, and is divided into three sections: EST human, EST mouse, and EST others. Even so, searches of these databases take far longer than a search of one of the non-redundant protein databases. You should only search an EST database if a search of a protein database has failed to find a match.
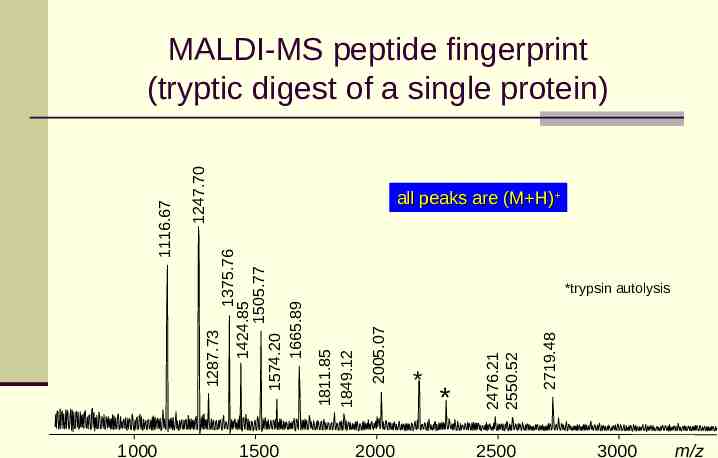
1247.70 1000 1500 2000 * 2500 2719.48 * 2476.21 2550.52 2005.07 *trypsin autolysis 1811.85 1849.12 1375.76 1424.85 1505.77 1574.20 1665.89 all peaks are (M H) 1287.73 1116.67 MALDI-MS peptide fingerprint (tryptic digest of a single protein) 3000 m/z
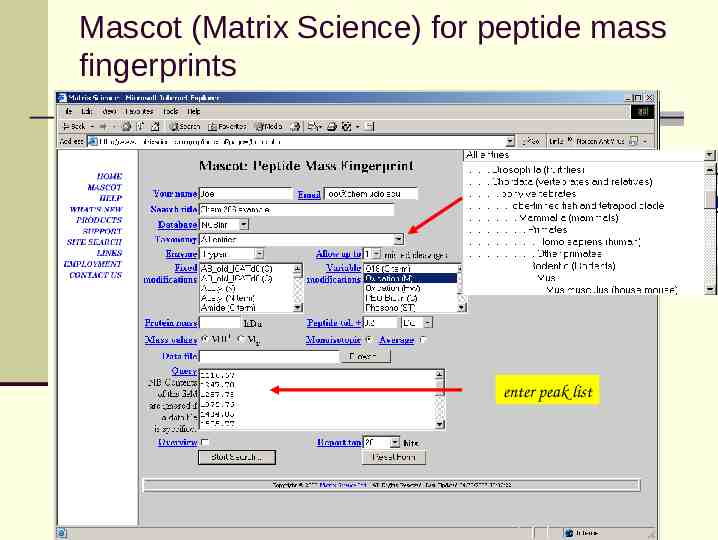
Mascot (Matrix Science) for peptide mass fingerprints enter peak list
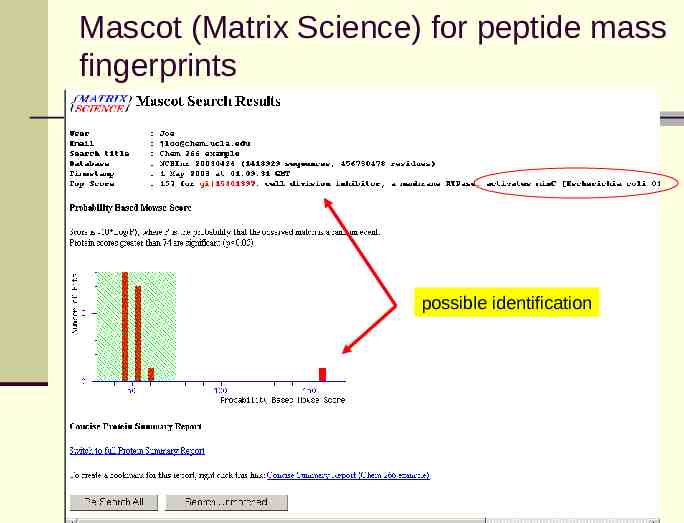
Mascot (Matrix Science) for peptide mass fingerprints possible identification
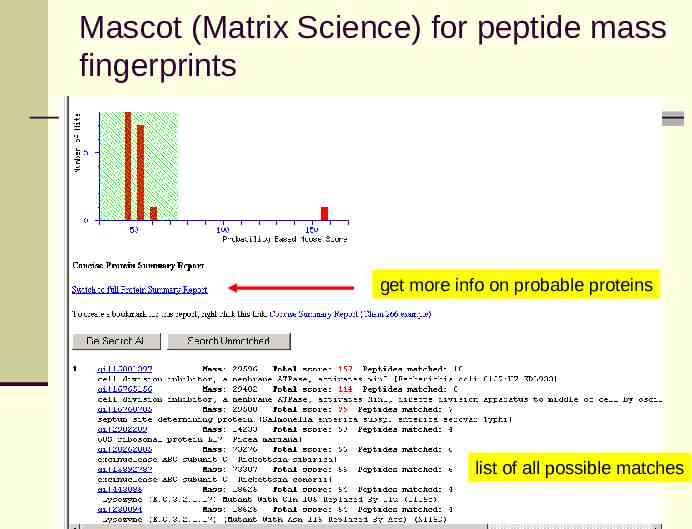
Mascot (Matrix Science) for peptide mass fingerprints get more info on probable proteins list of all possible matches
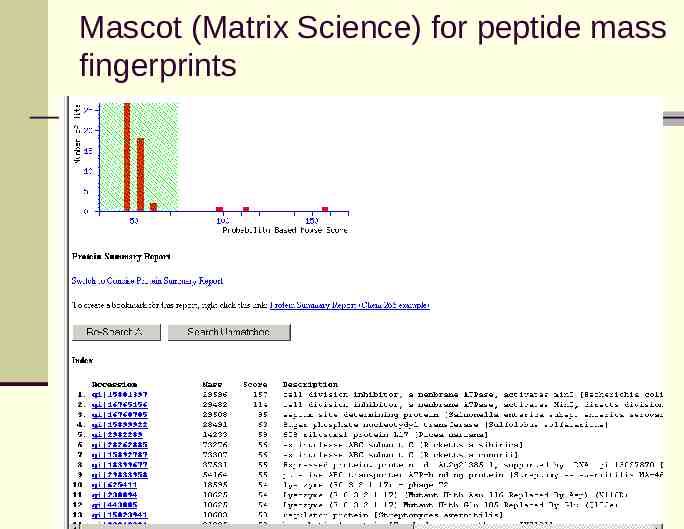
Mascot (Matrix Science) for peptide mass fingerprints
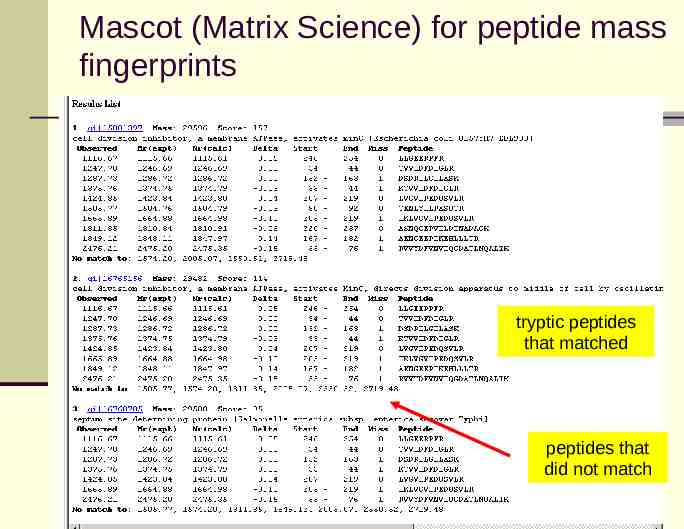
Mascot (Matrix Science) for peptide mass fingerprints tryptic peptides that matched peptides that did not match
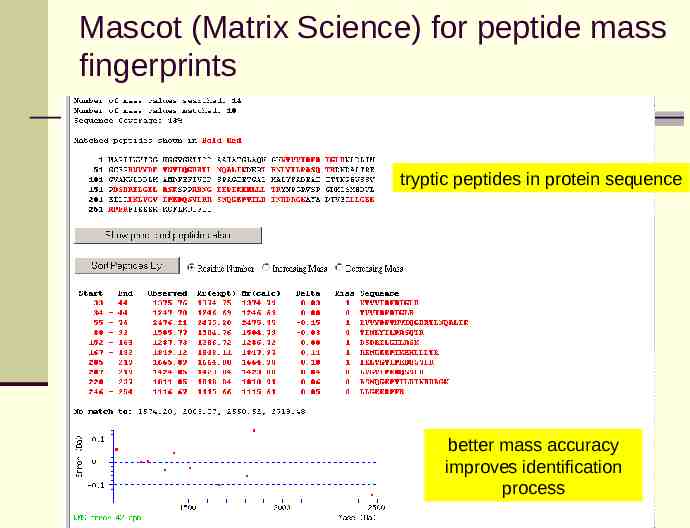
Mascot (Matrix Science) for peptide mass fingerprints tryptic peptides in protein sequence better mass accuracy improves identification process
LC-MS/MS for protein identification To provide further confirmation of the identification, if a tandem mass spectrometer (MS/MS) is available, peptide ions can be dissociated in the mass spectrometer to provide direct sequence information. Product ions from an MS/MS spectrum can be compared to available sequences using powerful software toolsl. For a single sample, LC-MS/MS analysis included two discrete steps: steps (a) LC-MS peptide mapping to identify peptide ions from the digestion mixture and to deduce their molecular weights, and (b) LC-MS/MS of the previously detected peptides to obtain sequence information for protein identification.
Automated LC-MS/MS and database searching Current mass spectral technology permits the generation of MS/MS data at an unprecedented rate. Prior to the generation of powerful computer-based database searching strategies, the largest bottleneck in protein identification was the manual interpretation of this MS/MS data to extract the sequence information. Today, many computer-based search strategies that employ MS/MS data require no operator interpretation at all. Analogous to the approach described for peptide fingerprinting, these programs take the individual protein entries in a database and electronically "digest" them to generate a list of theoretical peptides for each protein. However, in the use of MS/MS data, these theoretical peptides are further manipulated to generate a second level of lists which contain theoretical fragment ion masses that would be generated in the MS/MS experiment for each theoretical peptide.
Automated LC-MS/MS and database searching These programs simply compare the list of experimentally determined fragment ion masses from the MS/MS experiment of the peptide of interest with the theoretical fragment ion masses generated by the computer program. The recent advent of data-dependant scanning functions has permitted the unattended acquisition of MS/MS data. An example of a raw MS/MS data searching program that takes particular advantage of this ability is SEQUEST. SEQUEST will input the data from a data-dependant LC/MS chromatogram and automatically strip out all of the MS/MS information for each individual peak, and submit it for database searching using the strategy discussed above. Each peak is treated as a separate data file, making it especially useful for the on-line separation and identification of individual components in a protein mixture. SEQUEST cross-correlates uninterpreted MS/MS mass spectra of peptides from protein/nucleotide databases. The software can analyze a single spectrum or an entire LC-MS/MS peptide map. No user interpretation of MS/MS spectra is involved.
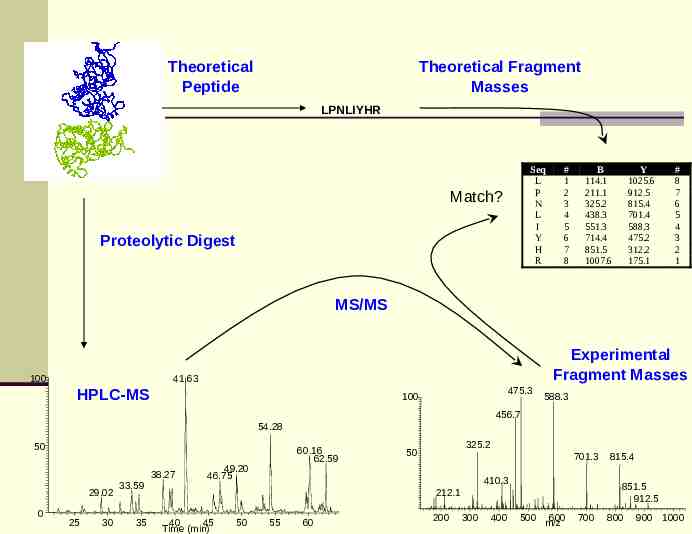
Theoretical Peptide Theoretical Fragment Masses LPNLIYHR Seq L P N L I Y H R Match? Proteolytic Digest # 1 2 3 4 5 6 7 8 B 114.1 211.1 325.2 438.3 551.3 714.4 851.5 1007.6 Y 1025.6 912.5 815.4 701.4 588.3 475.2 312.2 175.1 # 8 7 6 5 4 3 2 1 MS/MS 100 Experimental Fragment Masses 41.63 HPLC-MS 475.3 100 588.3 456.7 54.28 50 38.27 29.02 0 25 30 33.59 35 60.16 62.59 49.20 46.75 325.2 50 701.3 410.3 851.5 912.5 212.1 40 45 Time (min) 50 55 60 200 300 400 815.4 500 600 m/z 700 800 900 1000
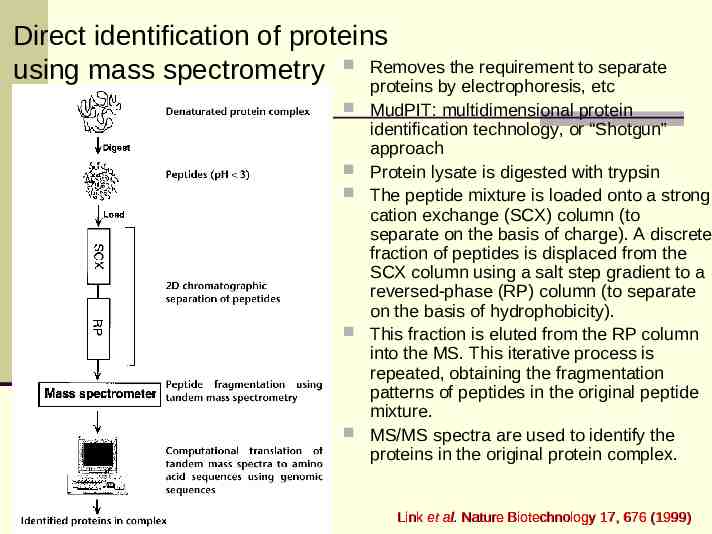
Direct identification of proteins the requirement to separate using mass spectrometry Removes proteins by electrophoresis, etc MudPIT: multidimensional protein identification technology, or “Shotgun” approach Protein lysate is digested with trypsin The peptide mixture is loaded onto a strong cation exchange (SCX) column (to separate on the basis of charge). A discrete fraction of peptides is displaced from the SCX column using a salt step gradient to a reversed-phase (RP) column (to separate on the basis of hydrophobicity). This fraction is eluted from the RP column into the MS. This iterative process is repeated, obtaining the fragmentation patterns of peptides in the original peptide mixture. MS/MS spectra are used to identify the proteins in the original protein complex. Link et al. Nature Biotechnology 17, 676 (1999)
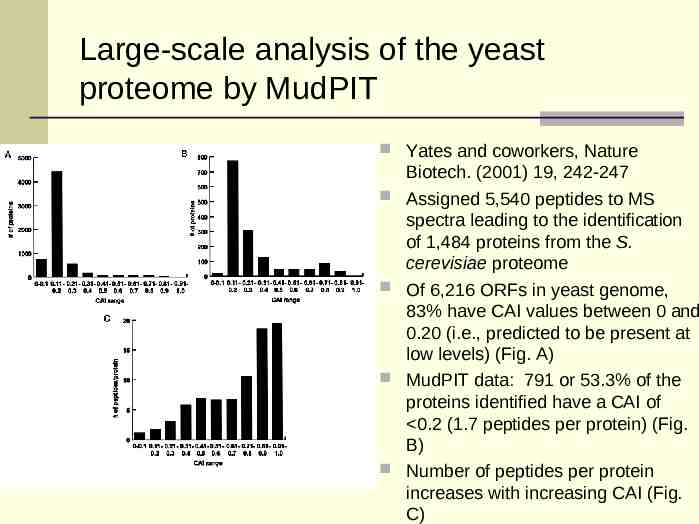
Large-scale analysis of the yeast proteome by MudPIT Yates and coworkers, Nature Biotech. (2001) 19, 242-247 Assigned 5,540 peptides to MS spectra leading to the identification of 1,484 proteins from the S. cerevisiae proteome Of 6,216 ORFs in yeast genome, 83% have CAI values between 0 and 0.20 (i.e., predicted to be present at low levels) (Fig. A) MudPIT data: 791 or 53.3% of the proteins identified have a CAI of 0.2 (1.7 peptides per protein) (Fig. B) Number of peptides per protein increases with increasing CAI (Fig. C)
Approaches for Protein Sequencing and Identification “Top Down” MS/MS MIRERICACVLALGMLTGFTHAFGSKDAAADGKPLVVTTIGMIADAVKNIAQGDVHLKGLMGP GVDPHLYTATAGDVEWLGNADLILYNGLHLETKMGEVFSKLRGSRLVVAVSETIPVSQRLSLE EAEFDPHVWFDVKLWSYSVKAVYESLCKLLPGKTREFTQRYQAYQQQLDKLDAYVRRKAQS LPAERRVLVTAHDAFGYFSRAYGFEVKGLQGVSTASEASAHDMQELAAFIAQRKLPAIFIESSI PHKNVEALRDAVQARGHVVQIGGELFSDAMGDAGTSEGTYVGMVTHNIDTIVAALAR The molecular mass of an intact protein defines the native covalent state of a gene’s product, including the effects of post-transcriptional/translational modifications, and associated heterogeneity, that are modulated by the actions of other gene products . Moreover, the fragmentation pattern from large proteins can generate sufficient information for identification from sequence databases, particularly when combined with accurate mass measurements of both the intact molecule and its product ions.
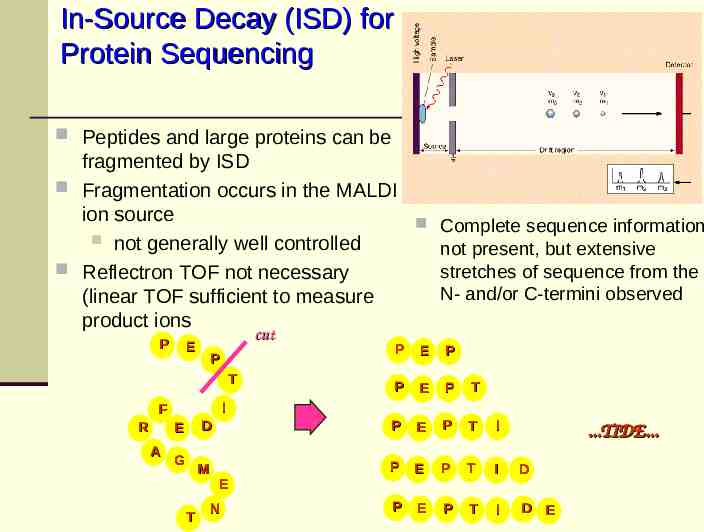
In-Source Decay (ISD) for Protein Sequencing Peptides and large proteins can be fragmented by ISD Fragmentation occurs in the MALDI ion source Complete sequence information not generally well controlled not present, but extensive Reflectron TOF not necessary stretches of sequence from the N- and/or C-termini observed (linear TOF sufficient to measure product ions P cut E P E P P E P T D P E P T I M P E P T I D P E P T I D E P T I F R E A G T E N .TIDE.
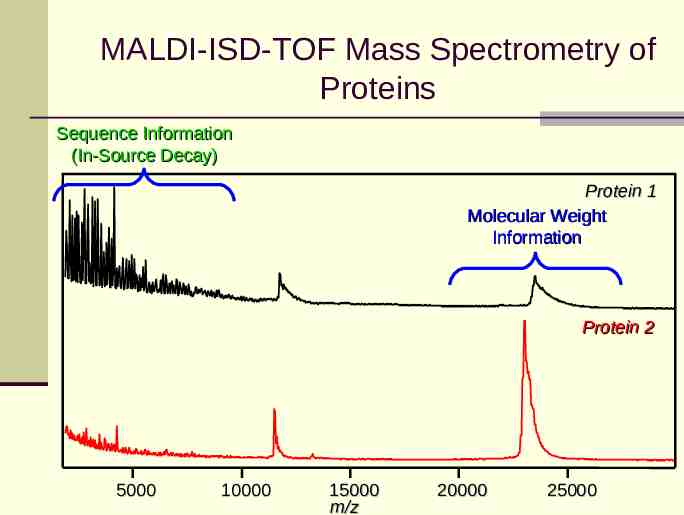
MALDI-ISD-TOF Mass Spectrometry of Proteins Sequence Information (In-Source Decay) Protein 1 Molecular Weight Information Protein 2 5000 10000 15000 m/z 20000 25000
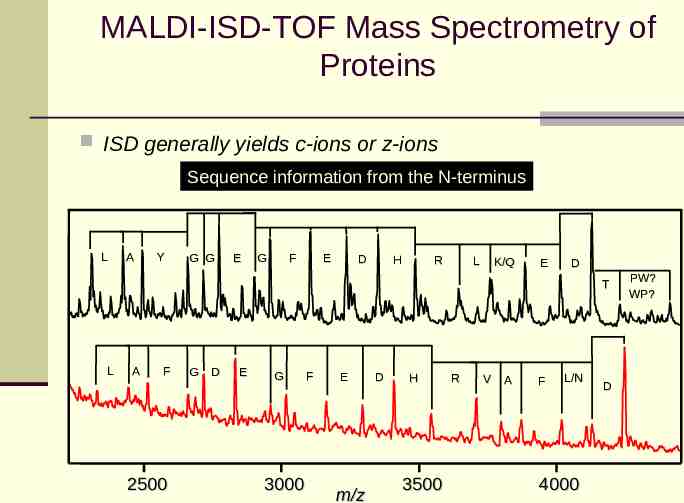
MALDI-ISD-TOF Mass Spectrometry of Proteins ISD generally yields c-ions or z-ions Sequence information from the N-terminus L A Y G G E G F E D H R L K/Q E D T L A F 2500 G D E G 3000 F E m/z D H 3500 R V A F L/N 4000 D PW? WP?
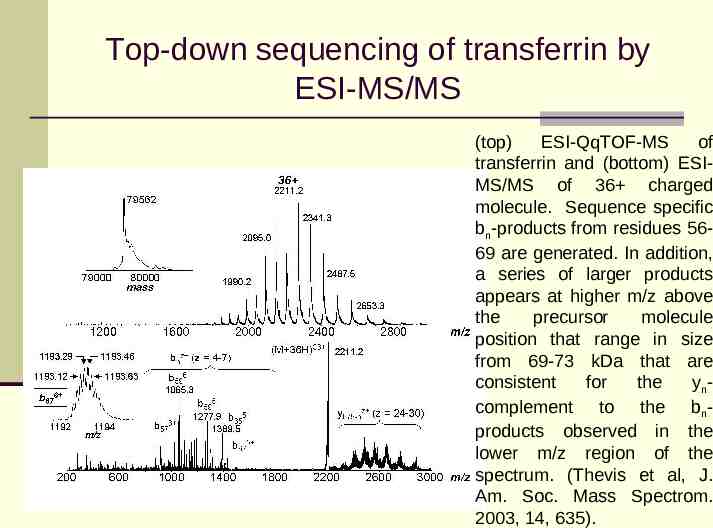
Top-down sequencing of transferrin by ESI-MS/MS (top) ESI-QqTOF-MS of transferrin and (bottom) ESIMS/MS of 36 charged molecule. Sequence specific bn-products from residues 5669 are generated. In addition, a series of larger products appears at higher m/z above the precursor molecule position that range in size from 69-73 kDa that are consistent for the yncomplement to the bnproducts observed in the lower m/z region of the spectrum. (Thevis et al, J. Am. Soc. Mass Spectrom. 2003, 14, 635).
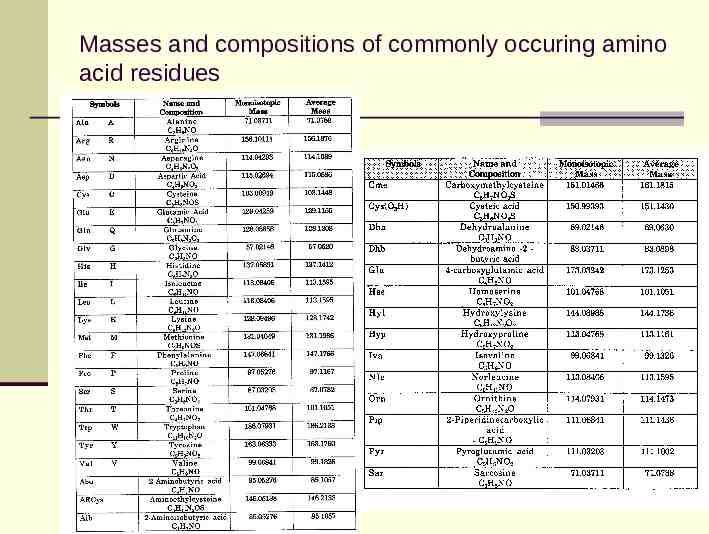
Masses and compositions of commonly occuring amino acid residues






