Chapter 4 Validation






















































56 Slides891.36 KB
Chapter 4 Validation
Chapter Objectives Know and use common validation terminology Understand how equipment, process, and method validation fits into the overall quality system Describe the types of validation documents found in a bio manufacturing organization, their typical content, and purpose Understand the validation life cycle Understand how a validation program is systematically established and the flow of validation requirements Distinguish procedures and outcomes for Design Qualification (DQ), Installation Qualification (IQ), Operation Qualification (OQ) and Performance Qualification (PQ)
What is Validation? FDA Definition: “The process of demonstrating, through documented evidence, that a process, procedure, piece of equipment, or analytical method, or facility will consistently produce a product or result that meets predetermined specifications and quality attributes”
Why is Validation Important in Bio manufacturing? Bio manufacturing is a complex process involving multiple unit operations many of which are critical to ensuring patient safety and product efficacy Validating the performance of unit operations, analytical methods, and critical process points ( sterilization, viral inactivation, cleaning procedures) is essential to ensure that the process generates a quality product Important part of the quality that must be built into the production process
Validation – Historical Basis The Cutter incident 1955, vaccine for polio was introduced, and over 10 million children in five countries were inoculated during the first year Cutter Laboratories – one of the companies recruited to produce the vaccine April 1955, California public health officials noticed an increase in cases of polio 200,000 people inoculated with a vaccine that contained live virus instead of an inactive virus! Company erroneously assumed rate of viral inactivation followed first order kinetics; actually was second order kinetics
Validation- Historical Basis Cont’d Large Volume Parenteral (LVP) Solutions that are injected into humans. Sterility of solution is critical 1971 – parenteral solutions used in a burn ward were incompletely sterilized contaminated with Pseudomonas spp. bacteria large number of patients came down with infections and over 50 died investigation discovered that positioning of materials in the autoclave the time and temperature used for sterilization, had not been tested on the actual material
FDA Response Expand requirements Good Manufacturing Practices (GMP) to include validation of equipment and processes 1976 – focus on various types of sterilization processes used in the pharmaceutical industry; and the need to validate those processes Filtration operations environmental controls water systems Aseptic processing operations
Validation in Biomanufacturing Essential part of GMPs, ensures that : the manufacturingfacility/system/equipment/metho d is consistently doing what is supposed to be doing (process is well controlled) decisions on processes and products are made based on good science and data - not assumptions process variables and acceptable limits for these variables and appropriate in-process controls are established
Validation in Biomanufacturing Cont’d Requires careful attention to raw material specifications, in process material specifications, and final product specifications Validation does not replace testing, but it does reduce the testing burden for raw materials, in-process materials, and final product
Validation Process Validation itself is a process that evolves with the product Validation requirements for production of pre-clinical material much less stringent then for phase III clinical material Critical operations: raw materials, analytical methods, viral clearance, sterilization, cleaning – these operations will require greater validation efforts then less critical operations (media blending)
Validation in Biomanufacturing A fully validated process is “locked in” Any change outside of the validated space invalidates process Change must be evaluated for effect on patient safety and product efficacy
Components Validation Plan Organization must define an approach towards validation What to validate How to validate Who is responsible for the validation Who approves the validation When validation and revalidation should be performed
Validation Plan Requirements Regulatory agencies (FDA, EMEA, WHO) require minimum components of validation Industry standards ( the “c” in cGMP) can increase validation requirements New and novel processes/equipment require greater scrutiny than established processes Validation requirements increase as product moves through development ( phase I, phase II, Phase III)
Validation Master Plan Validation Master Plan High level document that outlines the organizations approach to validation and revalidation. Guideline by which individual validation protocols are developed and implemented May contain a flow chart or other diagrams of the validation process
Validation Protocol Specific protocols (SOPs) that provide detailed information on what is to be validated. may consist of multiple SOP’s each describing specific steps in the validation process Includes: description of the process, equipment, or method to be validated\ description of the validation method description of the sampling procedure including type and number of samples acceptance criteria for test results schedule criteria for revalidation
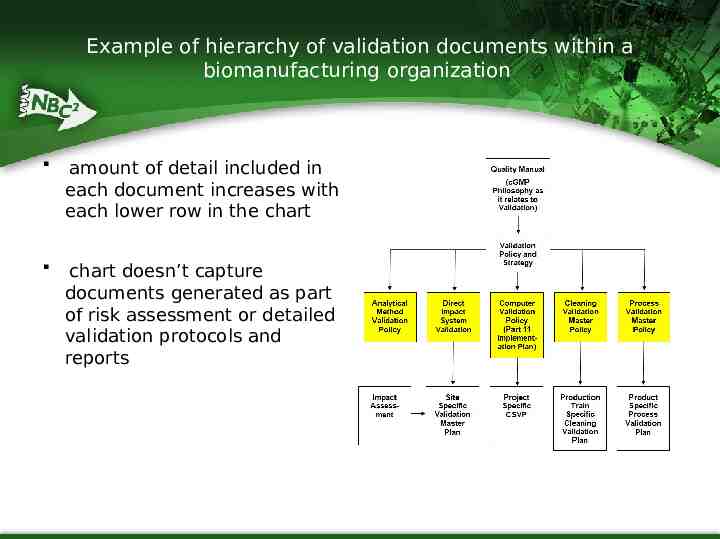
Example of hierarchy of validation documents within a biomanufacturing organization amount of detail included in each document increases with each lower row in the chart chart doesn’t capture documents generated as part of risk assessment or detailed validation protocols and reports
Critical Systems How critical is the system being validated to final product quality? Media blending systems for cell growth vs. final fill & finish operations Demonstrating that the device which fills, labels, and caps the final product will require more extensive validation then the blenders used to prepare media for bioreactors. Validation of complex devices can take years!
Systems Validation Examples of individual systems subject to validation: HVAC systems Autoclaves pH meters Depyrogenation Ovens Lyopholyzers Centrifuges Steam generators Water systems Compressed air systems Vacuum systems
Other Systems Also includes Analytical equipment and methods Analytical laboratory methods and equipment used in quality control process Computer systems/software validation Validation of systems of ensure accuracy, reliability, consistent intended performance, and the ability to discern invalid or altered records Cleaning validation Evaluate product residual reduction and detergent reduction to pre-determined acceptance limits
Validation Life Cycle Begins with the design of the manufacturing process Sustained through the use of equipment and trial production runs Culminating in a body of evidence that each piece of equipment, each analytical method, each step of the manufacturing process, and each production run will produce a consistent, quality product Continues through periodic re-validation at specified intervals or in the event of changes Maintenance of the “validated state”
Validation Life Cycle Cont’d Research & Development R&D Engineering Design Equipment Validation Process validation/Performance Qualification Revalidation Process Monitoring
Validation Life Cycle – R&D Develop and scale up the manufacturing process Establish critical process parameters
Engineering Design Stage Determine user requirements Develop specifications and drawings to met user requirements Review and select equipment and vendors Develop the validation master plan Assemble and build equipment according to GMP Complete start-up and commissioning
Design Stage Does the equipment meet the needs Is the autoclave big enough? Do you have the manuals, spare parts, can you plug it in? Is it installed properly (drain lines, vents, etc) Does it work? Does the autoclave reach the necessary temp. and pressure? Can the autoclave sterilize your equipment (worse case situation)? How does it work in the manufacturing process? Can it handle production quantities? Will failure compromise product quality?
Equipment Qualification Procedures to verify that equipment has been: Installed (IQ) and Operates (OQ) according to design specifications
IQ-OQ-PQ Installation Qualification (IQ) A process used to document that the piece of equipment was supplied and installed properly and that appropriate utilities, i.e., electrical, steam, gas, etc. are available to operate the equipment according to the manufacturers specifications. Operational Qualification (OQ) A process designed to supply the documented evidence that a piece of equipment operates as it is intended through all anticipated operational ranges. Performance (Process) Qualification (PQ) Verifies that a process / piece of equipment performs as it is intended to in the manufacturing process and produces product (in process or final) meeting predetermined specifications.
Installation Qualification (IQ) Answers the questions: Do you know what you have? Verified drawings Manufacturer’s documentation Critical components Do you know what it needs in order to work? Utilities Compressed air, Clean Steam, electric Environment Room Classification, Temperature, RH Workmanship Level, properly supported, adequate clearance Tied to Design Specs 27
Typical information in an IQ protocol Name and description of equipment, including model numbers Identification, including model and serial numbers Location of the equipment Any utility requirements, i.e. electrical voltage, steam or water pressure, etc. Any safety features of the equipment, including alarms, interlocks, or relief valves That all documentation, including manufacturers contact information, spare parts inventory, operational manual, and
Operational Qualification (OQ) Answers the question: Does the system or equipment work as advertised? Typical verifications: Flow rates Pressures Temperatures Valve sequencing Alarms and interlocks Tied to the manufacturer’s design and specifications (FRS)
OQ OQ demonstrates that the equipment or system operates as intended Typical OQ testing should include: documenting the version of the software that is being tested and any revisions to the software that occur during testing (e.g., changes made as a result of failed tests) sequence of operation testing (testing all control functionality of the equipment/system) alarm testing power outage testing Standard Operating Procedure and logbook verification (verify that the operational SOPs are indicative of operation of the equipment and system-redline document(s) as required)
Typical OQ Protocol Components Objective Responsibility Equipment required (Calibration verification & Traceability) SOP(s) used Equipment Identification Parameters measured (Specifications) Documentation
Autoclave OQ Document
Performance Qualification (PQ) Processes to verify that the process and or equipment is capable of performing reliably and consistently to produce a product to predetermined specifications
Performance Qualification (PQ) Does the system or equipment consistently meet your needs? Requires a clear statement of user requirements Verifies properly installed and operating equipment being operated by trained operators following approved procedures IQ and OQ complete Documented training Approved SOPs The same equipment may have more that one PQ, depending on the needs of different
Three A Magic Number? Requires at least 3 consistent runs “once miracle ; twice-coincidence; 3 times- science” Companies would strive to demonstrate that three batches of product through the manufacturing process demonstrated a validated process Is this a high degree of assurance? Increasingly, the process data determines validation Is variability low? Is the process well-controlled within acceptable limits?
(PQ) The PQ integrates procedures, personnel, systems, and materials to verify that the utility, environment, equipment, or support system produces the required output This output may be either a product contact utility (clean compressed air, purified water, etc.) or an environmental system such as HVAC Only “direct impact” systems will be subject to PQ
Validation of Analytical Methods and Equipment Validating the equipment used in the manufacturing process is a critical aspect of biopharmaceutical production No less important is validation of the analytical laboratory methods and equipment used in the QC process Analytical method validations are performed to confirm that the method(s) to be used on a qualified analytical instrument meet the specifications for the intended use
Analytical Method Qualification Qualification demonstrates that the method performs as expected before it is formally validated A qualified method should be capable of providing results consistent with proposed product specifications
Aspects of the Method For any analytical method there are several parameters that must be established for the method Selectivity or specificity Linearity- demonstration of the linear range or validation of curve-fitting algorithms Limit of Detection (LOD) Limit of Quantification (LOQ) Precision
Specificity The specificity of a method refers to the method’s ability to detect the analyte in question in the presence of similar compounds A method that has poor selectivity will give rise to frequent false positives, meaning the method falsely indicates that the substance being assessed is present
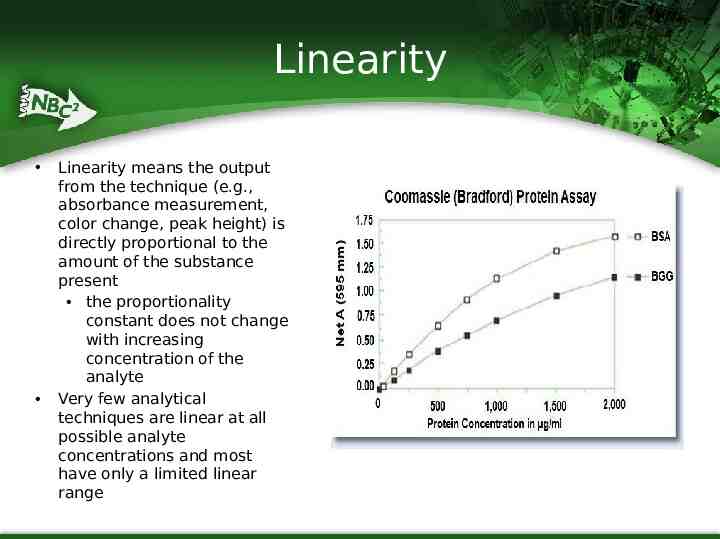
Linearity Linearity means the output from the technique (e.g., absorbance measurement, color change, peak height) is directly proportional to the amount of the substance present the proportionality constant does not change with increasing concentration of the analyte Very few analytical techniques are linear at all possible analyte concentrations and most have only a limited linear range
Methods at their Limits Limit of Detection (LOD) Establishes the lower limit at which we can detect the analyte of interest Useful in the measurement of impurities We cannot often say that a preparation is zero levels of some analyte but only that it contains less than the LOD Limit of Quantification (LOQ) Establishes the lower limit at which we can accurately quantify the analyte of interest The presence of the analyte might be detected at a lower level, but because of various background fluctuations (including instrument and reagent variability) it is possible that an accurate quantification of the analyte cannot be made
Precision Precision refers to the variability of test results when the test is performed on multiple portions, or aliquots, of a homogenous sample To separate or distinguish factors responsible for methods variation, regulatory guidance suggests breaking precision into areas that include repeatability (or method precision) and system precision
Computerized Systems and Software Validation Validating computerized systems can result in a very complex and involved validation effort CSV practices are not just internal to an organization—they extend to suppliers as well Suppliers must assure quality and integrity in all aspects of their product 44
Approach to Process Validation (FDA Guidance) Process Validation (PV) - the collection and evaluation of data, from the process design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering quality product Stage 1 – Process Design The commercial manufacturing process is defined during this stage based on knowledge gained through development and scale-up activities Stage 2 – Process Qualification During this stage, the process design is evaluated to determine if the process is capable of reproducible commercial manufacturing Stage 3 – Continued Process Verification Ongoing assurance is gained during routine production that the process remains in a state of control
Key Upstream Processes to Validate Cleaning Bioreactors, media formulation tanks, centrifuge Bioreactors, media, centrifuge Master and Working Cell Banks Fermentation/Cell Culture Process Product Recovery
Downstream Validation Protocols Scale-down qualification verify that the process operates the same at small-scale as it does at large-scale Column lifetime verify resin performance for maximum allowable duration and maximum number of process cycles Column cleaning verify cleaning procedures Buffer stability at all temperatures and vessels used in the process for maximum allowable hold duration Sanitization/storage buffers and verifying effectiveness Reprocessing (if applicable) verify consistency of product after reprocessing Small molecule clearance verify small molecule removal at each step Conformance purification and bulk fill verification demonstrate that the commercial process, when executed as specified in batch records, consistently produces in-process intermediates and Bulk Drug Substance (BDS) that meet all established specifications
Validation Ideally validation takes place prior to actual production runs, however in some cases validation may take place as product is produced, or past production runs may be used to provide validation data Prospective Validation Concurrent Validation Retrospective Validation
Revaildation/Process Monitoring Continually monitor the quality of the product and operational parameters Revalidate critical equipment (eg. autoclave, lyophilzer) routinely to show that validated state has been maintained Revalidate is any significant changes are made to the process or equipment is made
Revalidation Is the initial validation of a piece of equipment the end? No! Periodic revalidation may be necessary depending on the criticality of the equipment Changes need to be evaluated for their impact on validation Deviations from specifications may require revalidation Revalidation spelled out in Master Validation Plan
Change Control Must assess impact of changes on FDA compliance and validation state. Change control is a formal process defined in company SOP on how process/equipment changes are evaluated. Any change that takes place outside the change control process can jeopardize product quality (patient safety).
Change Control and Re-validation Change control – the process of evaluating operational changes must be controlled and evaluated for possible effects on product quality. Changes are defined as necessary to the continued operation of the validated system Unplanned – result of equipment failure or malfunction Planned – require a thorough assessment of the change on the process
Change Control and Re-validation Contd Document Change Order – updates to written procedures, particularly SOP’s Type of change requested Why the change is being requested Who is requesting the change Any regulatory impact from the change Quality Assurance decides if change is warranted
Validation Time consuming and expensive aspect, BUT essential to producing a safe and effective product Not a one and done event! Periodic revalidation is necessary to ensure the process, procedure, piece of equipment or analytical method continues to operate in a “validated state”
Risk-based Validation Risk analysis is a formal analytical activity Identify Assess Manage Risks considered in this process are related to Product Patient Employees of the biomanufacturer
Validation – Risk Based Approach improved from the old mindset “validate anything that moves and don’t move anything that is validated” current approach incorporates process understanding and risk assessment towards an elimination or reduction of those risks





