Basics of Rietveld Refinement Scott A Speakman 13-4009A






















25 Slides229.00 KB
Basics of Rietveld Refinement Scott A Speakman 13-4009A x3-6887 [email protected]
Uses of the Rietveld Method The Rietveld method refines user-selected parameters to minimize the difference between an experimental pattern (observed data) and a model based on the hypothesized crystal structure and instrumental parameters (calculated pattern) can refine information about a single crystal structure – confirm/disprove a hypothetical crystal structure – refine lattice parameters – refine atomic positions, fractional occupancy, and thermal parameter refine information about a single sample – preferred orientation refine information about a multiphase sample – determine the relative amounts of each phase Scott Speakman, 2007 Page 2
Requirements of Rietveld Method High quality experimental diffraction pattern a structure model that makes physical and chemical sense suitable peak and background functions Scott Speakman, 2007 Page 3
Obtaining High Quality Data issues to consider – aligned and calibrated instrument – beam overflow problems – thin specimen error – good counting statistics – appropriate step size – sample transparency – surface roughness – preferred orientation – particle size go to XRD Basics pg 102 Scott Speakman, 2007 Page 4
Describing the Crystal Structure space group lattice parameters atomic positions atomic site occupancies atomic thermal parameters – isotropic or anisotropic Scott Speakman, 2007 Page 5
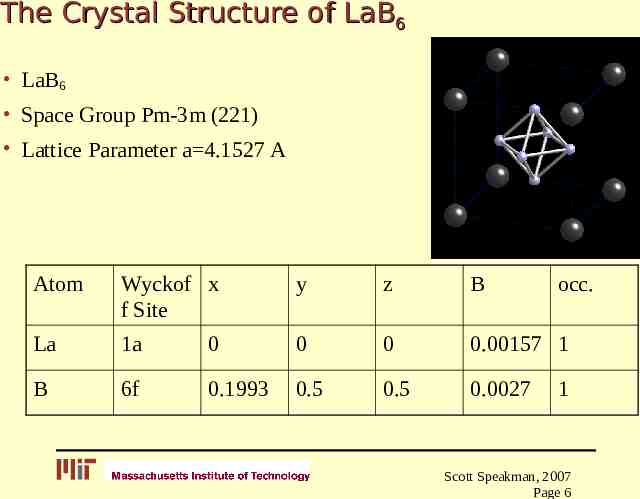
The Crystal Structure of LaB6 LaB6 Space Group Pm-3m (221) Lattice Parameter a 4.1527 A Atom Wyckof x f Site y z B occ. La 1a 0 0 0 0.00157 1 B 6f 0.1993 0.5 0.5 0.0027 1 Scott Speakman, 2007 Page 6
Where to get crystal structure information check if the structure is already solved – websites Inorganic Crystal Structure Database (ICSD) http://icsd.ill.fr/icsd/index.html 4% is available for free online as a demo Crystallography Open Database http://www.crystallography.net/ Mincryst http://database.iem.ac.ru/mincryst/index.php American Mineralogist http://www.minsocam.org/MSA/Crystal Database.html WebMineral http://www.webmineral.com/ – databases PDF4 from the ICDD Linus Pauling File from ASM International Cambridge Structure Database – literature use the PDF to search ICSD listings and follow the references look for similar, hopefully isostructural, materials index the cell, and then try direct methods or ab-initio solutions – beyond the scope of today’s class Scott Speakman, 2007 Page 7
Instrumental Parameters background peak profile parameters – cagliotti parameters u, v, w – pseudo-voigt or other profile parameters – asymmetry correction – anisotropic broadening error correcting parameters – zero shift – specimen displacement – absorption – extinction – roughness – porosity Scott Speakman, 2007 Page 8
How many parameters can we refine? Each diffraction peak acts as an observation – theoretically, refine n-1 parameters refining a tetragonal LaNi4.85Sn0.15 crystal structure, there might be: – scale factor – 2nd order polynomial background: 3 parameters – 2 lattice parameters – no atomic positions (all atoms are fixed) – 3 or 5 thermal parameters – 2 or 4 occupancy factors – zero shift and specimen displacement – 5 profile shape parameters 22 parameters maximum with 43 peaks (20 to 120 deg 2theta) – does this mean we can refine all parameters? Scott Speakman, 2007 Page 9
background functions manually fit background polynomial chebyshev shifte chebyshev amorphous sinc function many others for different programs Scott Speakman, 2007 Page 10
profile functions vary significantly with programs almost all programs use Cagglioti U, V, and W H 2 W V tan U tan 2 HSP uses pseudo-voigt, Pearson VII, Voigt, or pseudo-voigt 3 (FJC asymmetry) GSAS uses functions derived more from neutron and synchrotron beamlines Scott Speakman, 2007 Page 11
go to parameters calc pattern.pdf Scott Speakman, 2007 Page 12
How do you know if a fit is good? difference pattern Residuals R – R is the quantity that is minimized during least-squares or other fitting procedures Rwp wi Yio Yic i – Rwp is weighted to emphasize intense peaks over background – Rexp estimates the best value R for a data set an evaluation of how good the data are – RBragg tries to modify the R for a specific phase GOF (aka X2) Scott Speakman, 2007 Page 13 2
Refinement Strategy Rietveld methods fit a multivarialbe structure-backgroundprofile model to experimental data – lots of potential for false minima, diverging solutions, etc need to refine the most important variables first, then add more until an adequate solution is realized – a correct solution may not result Scott Speakman, 2007 Page 14
Ray Young’s Refinement Strategy scale factor zero shift or specimen displacement (not both) linear background lattice parameters more background peak width, w atom positions preferred orientation isotropic temperature factor B u, v, and other profile parameters anisotropic temperature factors Scott Speakman, 2007 Page 15
HSP Automatic Refinement Strategy Very similar to Prof Young’s recommendations a good choice for beginners you can set limits on any of these parameters Scott Speakman, 2007 Page 16
Additional Files XRD Basics HSP 2006.pdf – large collection of information about X-ray diffraction, instrumentation, and different techniques X’Pert HighScore Plus Tutorial.pdf – overview of the different functionality available in HighScore Plus Introduction.pdf – overview of Rietveld parameters calc patterns.pdf – overview of parameters involved in calculating a diffraction pattern Scott Speakman, 2007 Page 17
further reading “Rietveld refinement guidelines”, J. Appl.Cryst. 32 (1999) 36-50 R.A. Young (ed), The Rietveld Method, IUCr 1993 V.K. Pecharsky and P.Y. Zavalij, Fundamentals of Powder Diffraction and Structural Characterization of Materials, Kluwer Academic 2003. DL Bish and JE Post (eds), Modern Powder Diffraction, Reviews in Mineralogy vol 20, Min. Soc. Amer. 1989. CCP14 website http://www.ccp14.ac.uk/tutorial/tutorial.htm prism.mit.edu/xray/resources.htm Scott Speakman, 2007 Page 18
Rietveld Programs Free – – – – – – GSAS ExpGUI Fullprof Rietica PSSP (polymers) Maud (not very good) PowderCell (mostly for calculating patterns and transforming crystal structures, limited refinement) Commercial – PANalytical HighScore Plus – Bruker TOPAS (also an academic) – MDI Jade or Ruby Scott Speakman, 2007 Page 19
Examples Silicon LaB6 intermetallic LaNi4.85Sn0.15 Scott Speakman, 2007 Page 20
Silicon Open the datafile in HSP Add the structure model – insert the structure manually – import (insert) a struture file usually use the CIF format– the ubiquitous standard for crystal structures HSP can also import ICSD *.cry files and structures from other refinement programs GSAS can import CIF or PowderCell files try the automatic refinement manually improve the fit Scott Speakman, 2007 Page 21
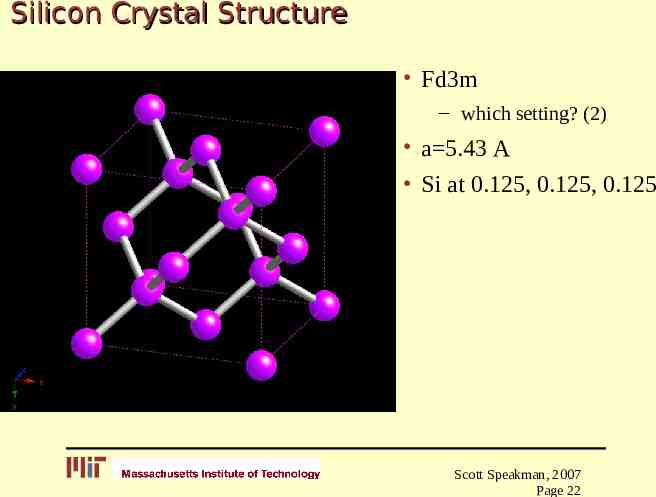
Silicon Crystal Structure Fd3m – which setting? (2) a 5.43 A Si at 0.125, 0.125, 0.125 Scott Speakman, 2007 Page 22
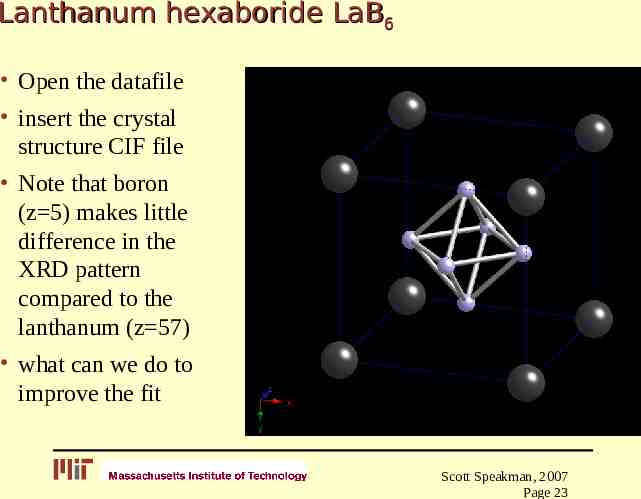
Lanthanum hexaboride LaB6 Open the datafile insert the crystal structure CIF file Note that boron (z 5) makes little difference in the XRD pattern compared to the lanthanum (z 57) what can we do to improve the fit Scott Speakman, 2007 Page 23
LaNi4.85Sn0.15 The data was taken from Chapter 6 of Fundamentals of Powder Diffraction and Structural Characterization of Materials, by Pecharsky and Zavalij The structure is a bit more complex that our earlier example, which allows us to explore more features of HighScore Plus The data (Ch6 1.raw) is in GSAS format, which can be read into HighScore Plus I have also included a CIF file from the ICSD (#104685) with all the main features of the structure described Scott Speakman, 2007 Page 24
Issue to Consider How can I work without knowledge of the structure? – Use LeBail or Pawley method to determine lattice parameters – Try indexing and solving the structure using the HighScore Plus tools – You will find that there are 16 possible space groups for this material, but picking the most common (and simplest) choice, P6/mmm, is the right way to go Where do I put the atoms? – You can use a Fourier map to find out wherein the structure the electron densities are greatest. Put the heaviest atoms (La) at these sites, then work your way through the chemistry What variables do I refine and in what sequence? – Take a look at the “automatic” option in HSP - this is not a bad strategy to use. We will go through these in detail Scott Speakman, 2007 Page 25






